Essential Requirements Checklist of the MDD(07 47 EC)基本要求检查表(中英文)
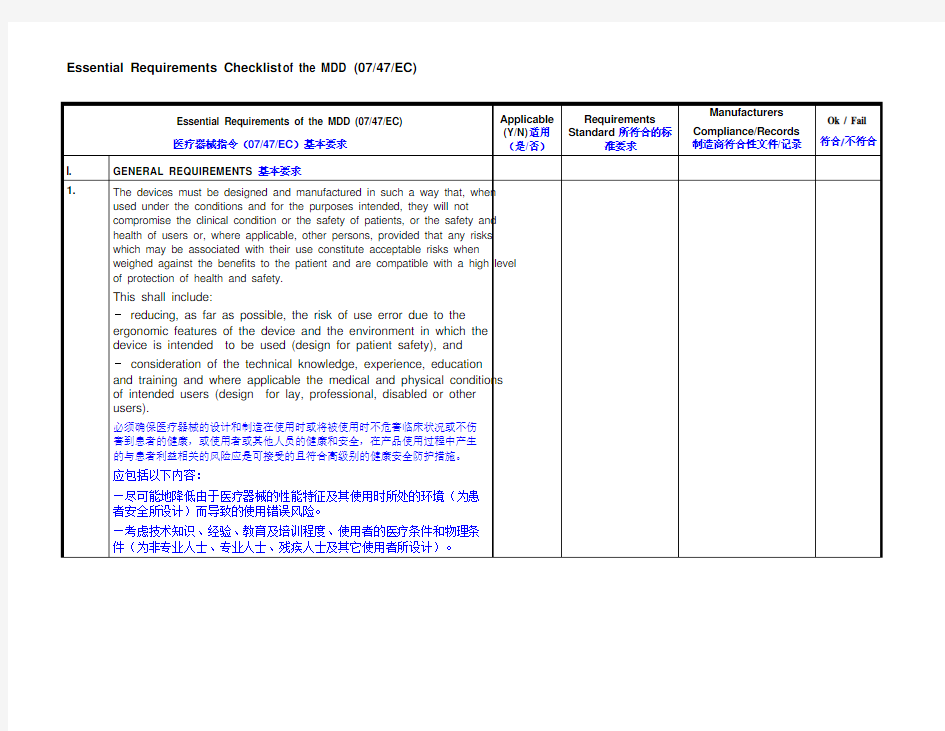

Essential Requirements of the MDD (07/47/EC) 医疗器械指令(07/47/EC)基本要求Applicable
(Y/N)适用
(是/否)
Requirements
Standard 所符合的标
准要求
Manufacturers
Compliance/Records
制造商符合性文件/记录
Ok / Fail
符合/不符合
I. GENERAL REQUIREMENTS基本要求
1.The devices must be designed and manufactured in such a way that, when
used under the conditions and for the purposes intended, they will not
compromise the clinical condition or the safety of patients, or the safety and
health of users or, where applicable, other persons, provided that any risks
which may be associated with their use constitute acceptable risks when
weighed against the benefits to the patient and are compatible with a high level
of protection of health and safety.
This shall include:
- reducing, as far as possible, the risk of use error due to the
ergonomic features of the device and the environment in which the
device is intended to be used (design for patient safety), and
- consideration of the technical knowledge, experience, education
and training and where applicable the medical and physical conditions
of intended users (design for lay, professional, disabled or other
users).
必须确保医疗器械的设计和制造在使用时或将被使用时不危害临床状况或不伤
害到患者的健康,或使用者或其他人员的健康和安全,在产品使用过程中产生
的与患者利益相关的风险应是可接受的且符合高级别的健康安全防护措施。
应包括以下内容:
—尽可能地降低由于医疗器械的性能特征及其使用时所处的环境(为患
者安全所设计)而导致的使用错误风险。
—考虑技术知识、经验、教育及培训程度、使用者的医疗条件和物理条
件(为非专业人士、专业人士、残疾人士及其它使用者所设计)。
Essential Requirements of the MDD (07/47/EC) 医疗器械指令(07/47/EC)基本要求Applicable
(Y/N)适用
(是/否)
Requirements
Standard 所符合的标
准要求
Manufacturers
Compliance/Records
制造商符合性文件/记录
Ok / Fail
符合/不符合
2.The solutions adopted by the manufacturer for the design and construction of
the devices must conform to safety principles, taking account of the generally
acknowledged state of the art.
In selecting the most appropriate solutions, the manufacturer must apply the
following principles in the following order:
制造商所采用的医疗器械的设计方法和结构方法必须符合安全准则,符合大众
公认的技术声明文件。
制造商必须依次采用以下准则以选择最适合的方法:
?eliminate or reduce risks as far as possible (inherently safe design and construction),
?尽可能地消除或降低风险(固有安全设计和结构)
?where appropriate take adequate protection measures including alarms if necessary, in relation to risks that cannot be eliminated,
?有关风险不能被消除时,应采取适当的防护措施,必要时包括安装警报装置。
?inform users of the residual risks due to any shortcomings of the protection measures adopted.
?把采取了安全措施后仍然可能存在的风险告知给使用者。
3.The devices must achieve the performances intended by the manufacturer and
be designed, manufactured and packaged in such a way that they are suitable
for one or more of the functions referred to in Article 1 (2) (a), as specified by
the manufacturer.
制造商所设计的医疗器械必须达到预期的性能,并且其设计、生产和包装必须
符合条款1(2)(a),由制造商规定的至少一项功能。
Essential Requirements of the MDD (07/47/EC) 医疗器械指令(07/47/EC)基本要求Applicable
(Y/N)适用
(是/否)
Requirements
Standard 所符合的标
准要求
Manufacturers
Compliance/Records
制造商符合性文件/记录
Ok / Fail
符合/不符合
4. The characteristics and performances referred to in sections 1, 2 and 3 must
not be adversely affected to such a degree that the clinical condition and
safety of the patients and, where applicable, of other persons are
compromised during the lifetime of the device as indicated by the
manufacturer, when the device is subjected to the stresses which can occur
during normal conditions of use.涉及条款1,2和3的产品的特征和性能在制造
商所指明的产品有效期内、在正常使用条件下受到挤压时必须不影响临床状
况、患者安全以及其他人员的安全。
5.The devices must be designed, manufactured and packed in such a way that
their characteristics and performances during their intended use will not be
adversely affected during transport and storage taking account of the
instructions and information provided by the manufacturer
医疗器械的设计、制造和包装在其预期使用过程中其特性和性能不应受
到损害,在按制造商提供的说明书和资料进行运输和储存时要保证其特
性和性能的完整性。
6.Any undesirable side effects must constitute an acceptable risk when weighed
against the performances intended.
6a. Demonstration of conformity with the essential requirements must
include a clinical evaluation in accordance with Annex X.
当医疗器械的预期性能受到影响时,任何不良副作用必须是可接受的风险。
6a.依据附录X,符合性声明和基本要求中必须含有临床评估。
II. REQUIREMENTS REGARDING DESIGN AND CONSTRUCTION
设计和结构要求
7.Chemical, physical and biological properties
化学性能、物理性能和生物性能
7.1 The devices must be designed and manufactured in such a way as to
guarantee the characteristics and performances referred to in Section 1 on the
"General requirements". Particular attention must be paid to:
医疗器械的设计和制造必须确保其符合“一般要求”中条款1所描述的特征和
性能。应特别注意:
Essential Requirements of the MDD (07/47/EC) 医疗器械指令(07/47/EC)基本要求Applicable
(Y/N)适用
(是/否)
Requirements
Standard 所符合的标
准要求
Manufacturers
Compliance/Records
制造商符合性文件/记录
Ok / Fail
符合/不符合
?the choice of materials used, particularly as regards toxicity and, where appropriate flammability,
?所用物料的选择,尤其要注意其毒性,必要时还要考虑其可燃性。
?the compatibility between the materials used and biological tissues, cells and body fluids, taking account of the intended purpose of the device.
?where appropriate, the results of biophysical or modelling research whose validity has been demonstrated beforehand.
?根据医疗器械的预期用途,确保其所使用的物料与生物组织、细胞和体液之间的兼容性。
?必要时,生物或模化研究结果的有效性事先应得到验证。
7.2 The devices must be designed, manufactured and packed in such a way as to
minimise the risk posed by contaminants and residues to the persons involved
in the transport, storage and use of the devices and to the patients, taking
account of the intended purpose of the product. Particular attention must be
paid to the tissues exposed and the duration and frequency of the exposure.
考虑到医疗器械的预期用途,医疗器械的设计、制造和包装必须使污染物和残
余物质对在运输、存储和使用过程中所涉及到的人员和患者所造成的风险降低
到最低。尤其要注意暴露组织、持续时间和暴露频率。
7.3 The devices must be designed and manufactured in such a way that they can
be used safely with the materials, substances and gases with which they enter
into contact during their normal use or during routine procedures; if the devices
are intended to administer medicinal products they must be designed and
manufactured in such a way as to be compatible with the medicinal products
concerned according to the provisions and restrictions governing those
products and that their performance is maintained in accordance with the
intended use.
医疗器械的设计和制造必须确保其所使用的物料、物质和气体在正常使用或惯
常使用情况下能安全使用。如果医疗器械用于施用医疗产品,其设计和制造必
须符合医疗的相关条款和制度,并按照预期用途保持其性能。
Essential Requirements of the MDD (07/47/EC) 医疗器械指令(07/47/EC)基本要求Applicable
(Y/N)适用
(是/否)
Requirements
Standard 所符合的标
准要求
Manufacturers
Compliance/Records
制造商符合性文件/记录
Ok / Fail
符合/不符合
7.4 Where a device incorporates, as an integral part, a substance which, if
used separately, may be considered to be a medicinal product as
defined in Article 1 of Directive 2001/83/EC and which is liable to act
upon the body with action ancillary to that of the device, the quality,
safety and usefulness of the substance must be verified by analogy
with the methods specified in Annex I to Directive 2001/83/ EC.
当医疗器械组合成一个整体部件,其组合所使用的物质在单独使用时按
照2001/83EC指令中章节1的规定可视作医疗产品,当作用于人体
时,该物质在医疗器械组合部件中起辅助作用,必须根据2001/83/EC
指令附录 1中规定的方法类似的方法对物质的质量、安全和效用进行验
证。
For the substances referred to in the first paragraph, the notified body
shall, having verified the usefulness of the substance as part of the
medical device and taking account of the intended purpose of the
device, seek a scientific opinion from one of the competent authorities
designated by the Member States or the European Medicines Agency
(EMEA) acting particularly through its committee in accordance with
Regulation (EC) No 726/2004 (1) on the quality and safety of the
substance including the clinical benefit/risk profile of the incorporation
of the substance into the device.
关于第一段中所提到的物质,根据第726/2004号规则(1)中的规定,
报告主体应对视作医疗产品组成部分的物质的效用进行验证并考虑到医
疗器械的预期用途,关于物质的质量和安全,包括物质组合成医疗器械
后产生的临床效益和风险,应通过其委员会寻求来自成员国或欧洲医疗
代理机构指定的权威机构的科学观点。
When issuing its opinion, the competent authority or the EMEA shall
take into account the manufacturing process and the data related to
the usefulness of incorporation of the substance into the device as
determined by the notified body.
当提出观点时,权威机构或欧洲医疗代理机构应把报告主体所规定的制
造过程和物质组合成医疗器械后的相关效用数据考虑在内。
Essential Requirements of the MDD (07/47/EC) 医疗器械指令(07/47/EC)基本要求Applicable
(Y/N)适用
(是/否)
Requirements
Standard 所符合的标
准要求
Manufacturers
Compliance/Records
制造商符合性文件/记录
Ok / Fail
符合/不符合
Where a device incorporates, as an integral part, a human blood derivative, the notified body shall, having verified the usefulness of the substance as part of the medical device and taking into account the intended purpose of the device, seek a scientific opinion from the EMEA, acting particularly through its committee, on the quality and safety of the substance including the clinical benefit/risk profile of the incorporation of the human blood derivative into the device.
当医疗器械组合成一个整体部件,一个人体血液衍生物,报告主体应对视作医疗产品组成部分的物质的效用进行验证并考虑到医疗器械的预期用途,关于物质的质量和安全,包括物质组合成医疗器械后产生的临床效益和风险,应通过其委员会寻求来自成员国或欧洲医疗代理机构指定的权威机构的科学观点。
When issuing its opinion, the EMEA shall take into account the manufacturing process and the data related to the usefulness of incorporation of the substance into the device as determined by the notified body.
当提出观点时,权威机构或欧洲医疗代理机构应把报告主体所规定的制造过程和物质组合成医疗器械后的相关效用数据考虑在内。
Essential Requirements of the MDD (07/47/EC) 医疗器械指令(07/47/EC)基本要求Applicable
(Y/N)适用
(是/否)
Requirements
Standard 所符合的标
准要求
Manufacturers
Compliance/Records
制造商符合性文件/记录
Ok / Fail
符合/不符合
Where changes are made to an ancillary substance incorporated in a device, in particular related to its manufacturing process, the notified body shall be informed of the changes and shall consult the relevant medicines competent authority (i.e. the one involved in the initial consultation) , in order to confirm that the quality and safety of the ancillary substance are maintained.
当组合到医疗器械的辅助性物质发生变化时,特别是与之相关的制造过程发生变化时,应把这些变化通知到报告主体且咨询相关医疗权威机构(例如首次咨询的机构),以确保辅助性物质的质量和安全得到保持。The competent authority shall take into account the data related to the usefulness of incorporation of the substance into the device as determined by the notified body, in
(*)Regulation (EC) No 726/2004 of the European Parliament and of the Council of 31 March 2004 laying down Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency (OJ L 136, 30.4.2004, p. 1) . Regulation as last amended by Regulation (EC) No 1901/2006.
根据欧洲议会制定的第726/2004号(EC)规定第(*)条款和和欧洲委员会2004年3月31日制定的关于用于人类和动物的医疗产品的授权和监督程序以及建立欧洲医疗代理机构(OJL 136,30.4.2004,p.1)的决议,权威机构应把报告主体所规定的制造过程和物质组合成医疗器械后的相关效用数据考虑在内。
Essential Requirements of the MDD (07/47/EC) 医疗器械指令(07/47/EC)基本要求Applicable
(Y/N)适用
(是/否)
Requirements
Standard 所符合的标
准要求
Manufacturers
Compliance/Records
制造商符合性文件/记录
Ok / Fail
符合/不符合
7.5 The devices must be designed and manufactured in such a way as to
reduce to a minimum the risks posed by substances leaking from the
device. Special attention shall be given to substances which are
carcinogenic, mutagenic or toxic to reproduction, in accordance with
Annex I to Council Directive 67/548/EEC of 27 June 1967 on the
approximation of laws, regulations and administrative provisions
relating to the classification, packaging and labelling of dangerous
substances (*) . If parts of a device (or a device itself) intended to
administer and/or remove medicines, body liquids or other substances
to or from the body, or devices intended for transport and storage of
such body fluids or substances, contain phthalates which are classified
as carcinogenic, mutagenic or toxic to reproduction, of category 1 or 2,
in accordance with Annex I to Directive 67/548/EEC, these devices
must be labelled on the device itself and/or on the packaging for each
unit or, where appropriate, on the sales packaging as a device
containing phthalates.医疗器械的设计和制造必须确保由于医疗器械的
物质泄漏所引起的风险降低到最小值。根据1967年6月27日制定的
67/548/EEC指令附录1中的规定以及其它相关的法规、规章和管理规
定中关于危险物质的分类、包装和贴标,要特别注意致癌物质、诱导有
机体突变的物质或影响生殖的有毒物质。当医疗器械的部件(或医疗器
械本身)用于向人体施用和/或移除来自人体的药物、体液或其它物质,
或用于运输和存储这些体液或物质,包括根据67/548/EEC指令附录I
中目录1或目录2被归类为致癌物质、诱导有机体突变的物质或影响生
殖的有毒物质的邻苯二甲酸盐,必须在医疗器械产品上和/或在每一个包
装上或必要时在其销售包装上贴上标签,表明其含有邻苯二甲酸盐。
Essential Requirements of the MDD (07/47/EC) 医疗器械指令(07/47/EC)基本要求Applicable
(Y/N)适用
(是/否)
Requirements
Standard 所符合的标
准要求
Manufacturers
Compliance/Records
制造商符合性文件/记录
Ok / Fail
符合/不符合
If the intended use of such devices includes treatment of children or
treatment of pregnant or nursing women, the manufacturer must
provide a specific justification for the use of these substances with
regard to compliance with the essential requirements, in particular of
this paragraph, within the technical documentation and, within the
instructions for use, information on residual risks for these patient
groups and, if applicable, on appropriate precautionary measures
当医疗器械的使用对象包括儿童或孕妇或哺乳妇女,制造商必须提供使
用这些物质的详细理由及其所符合的基本要求,特别是本节要求,所要
提供的资料包括技术性文件、使用说明书、残余风险相关信息,若可
以,应提供相关预防措施。
7.6 The devices must be designed and manufactured in such a way as to reduce
as much as possible, risks posed by the unintentional ingress of substances
into the device taking into account the device and the nature of the
environment in which it is intended to be used.
考虑到医疗器械及其预期使用的环境特性,医疗器械的设计和制造必须尽可能
的降低由于外来物质进入医疗器械所造成的风险。
8. Infection and microbial contamination病毒传染和微生物污染
8.1 The devices and their manufacturing processes must be designed in such a
way as to eliminate or reduce as far as possible the risk of infection to the
patient, user and third parties. The design must allow easy handling and,
where necessary, minimise contamination of the device by the patient or vice
versa during use.
医疗器械及其生产过程的设计必须尽可能的消除或降低患者、使用者和第三方
被传染的风险。医疗器械的设计必须易于操作,必要时,降低医疗器械在使用
过程中被患者污染的程度或医疗器械对患者的污染程度。
8.2 Tissues of animal origin must originate from animals that have been subjected
to veterinary controls and surveillance adapted to the intended use of the
tissues.动物组织必须来源于组织的预期用途由兽医控制和监督的动物。
Notified Bodies shall retain information on the geographical origin of the
animals.认证主体应保留动物原产地相关信息。
Essential Requirements of the MDD (07/47/EC) 医疗器械指令(07/47/EC)基本要求Applicable
(Y/N)适用
(是/否)
Requirements
Standard 所符合的标
准要求
Manufacturers
Compliance/Records
制造商符合性文件/记录
Ok / Fail
符合/不符合
Processing, preservation, testing and handling of tissues, cells and substances
of animal origin must be carried out so as to provide optimal security. In
particular safety with regard to viruses and other transferable agents must be
addressed by implementation of validated methods of elimination or viral
inactivation in the course of the manufacturing process.对动物组织、细胞和物
质进行加工、存储、测试和处理时必须能够确保其最佳安全性。特别是在生产
加工过程中,必须通过执行有效的根除或灭菌方法对病毒和其它可传播媒介进
行处理。
8.3 Devices delivered in a sterile state must be designed, manufactured and
packed in a non-reusable pack and/or according to appropriate procedures to
ensure they are sterile when placed on the market and remain sterile, under
the storage and transport conditions laid down, until the protective packaging
is damaged or opened.以无菌形式提供的医疗器械的设计、制造和包装必须采
用一次性包装,并且/或根据适当的程序确保其在规定的存储和运输条件下在上
市时是无菌的并继续有效,直到其包装受损或被拆开。
8.4 Devices delivered in a sterile state must have been manufactured and
sterilised by an appropriate, validated method.以无菌形式提供的医疗器械必须
采用适当的、经认可的方法进行生产和灭菌。
8.5 Devices intended to be sterilised must be manufactured in appropriately
controlled (e.g. environmental) conditions.无菌医疗器械的制造必须在有适当控
制的(例如:环境控制)条件中进行。
8.6 Packaging systems for non-sterile devices must keep the product without
deterioration at the level of cleanliness stipulated and, if the devices are to be
sterilised prior to use, minimise the risk of microbial contamination. The
packaging system must be suitable taking account of the method of
sterilisation indicated by the manufacturer.非无菌医疗器械的包装系统必须确保
产品在所规定的洁净水平下没有变质,医疗器械在使用前要消毒以降低微生物
污染风险。包装系统必须适当考虑制造商所指定的消毒方法。
8.7 The packaging and/or label of the device must distinguish between identical or
similar products sold in both sterile and non-sterile condition.包装和/或标签必
须能够区分以无菌状态或非无菌状态销售的同类产品或相似产品。
9.Construction and environmental properties结构和环境特性
Essential Requirements of the MDD (07/47/EC) 医疗器械指令(07/47/EC)基本要求Applicable
(Y/N)适用
(是/否)
Requirements
Standard 所符合的标
准要求
Manufacturers
Compliance/Records
制造商符合性文件/记录
Ok / Fail
符合/不符合
9.1 If the device is intended for use in combination with other devices or
equipment, the whole combination, including the connection system must be
safe and must not impair the specified performance of the devices. Any
restrictions on use must be indicated on the label or in the instruction for use.
如果医疗器械与其它医疗器械或仪器结合使用,那么整个组合体、包括连接系
统必须安全且不损害该医疗器械的特有性能。任何使用限制必须在标签或使用
说明书中标明。
9.2 Devices must be designed and manufactured in such a way as to remove or
minimise as far as possible:
医疗器械的设计和制造必须尽可能消除或减少:
?the risk of injury, in connection with their physical features, including the volume/pressure ratio, dimensional, and where appropriate the ergonomic
features,
?由其物理特性,包括体积/压力,尺寸,必要时还包括人体工程特性所引起的受伤风险。
?risks connected with reasonably foreseeable environmental conditions, such as magnetic fields, external electrical influences, electrostatic
discharge, pressure, temperature or variations in pressure, and
acceleration,
?与一定的可预见性环境条件,例如磁场、外部电力影响,静电放电,气压,温度或压力变化和加速度有关的风险。
?the risks of reciprocal interference with other devices normally used in the investigations or for the treatment given,
?与通常用于研究或治疗的其它医疗器械发生相互干扰的风险。
?risks arising where maintenance or calibration are not possible (as with implants) from ageing of the materials used or loss of accuracy of any
measuring or control mechanism.
?在不可能进行维修保养或校准(当移植时)时,由于所使用的物料老化或任何测量失准或操作不当所产生的风险。
Essential Requirements of the MDD (07/47/EC) 医疗器械指令(07/47/EC)基本要求Applicable
(Y/N)适用
(是/否)
Requirements
Standard 所符合的标
准要求
Manufacturers
Compliance/Records
制造商符合性文件/记录
Ok / Fail
符合/不符合
9.3 Devices must be designed and manufactured in such a way as to minimise the
risks of fire or explosion during normal use and in single fault condition.
Particular attention must be paid to devices whose intended use includes
exposure to flammable substances which could cause combustion.
医疗器械的设计和制造必须能够降低在其正常使用过程中和出现单一故障时所
产生的火灾风险或爆炸风险。应特别注意含有在使用时由于曝光而可能引起燃
烧的易燃物质的医疗器械。
10.Devices with a measuring function具有测量功能的医疗器械
10.1 Devices with a measuring function must be designed and manufactured in
such a way as to provide sufficient accuracy and stability within appropriate
limits of accuracy and taking account of the intended purpose of the device.
The limits of accuracy must be indicated by the manufacturer.
具有测量功能的医疗器械的设计和制造应考虑其预期用途,并确保其在适当的
精确限制值范围内具有高精确度和稳定性。制造商必须指明精确度范围。
10.2 The measurement, monitoring and display scale must be designed in line with
ergonomic principles, taking account of the intended purpose of the device.
考虑到医疗器械的预期用途,测量、监测和显示刻度的设计必须符合人类工程
学原理。
10.3 The measurements made by devices with a measuring function must be
expressed in legal units conforming to the provisions of Council Directive
80/181/EEC (OJ No L 39, 15. 2. 1980, p. 40. Directive as last amended by
Directive 89/617/EEC (OJ No L 357, 7. 12. 1989, p. 28)).
具有测量功能的医疗器械的测量必须用 80/181/EEC 指令(OJ No L 39, 15. 2.
1980, p. 40. 由 89/617/EEC 指令(OJ No L 357, 7. 12. 1989, p. 28))修订而来)
所规定的法定单位表示。
11.Protection against radiation辐射防护
Essential Requirements of the MDD (07/47/EC) 医疗器械指令(07/47/EC)基本要求Applicable
(Y/N)适用
(是/否)
Requirements
Standard 所符合的标
准要求
Manufacturers
Compliance/Records
制造商符合性文件/记录
Ok / Fail
符合/不符合
11.1 11.1.1 General总则
Devices shall be designed and manufactured such that exposure of patients, users and other persons to radiation shall be reduced as far as possible compatible with the intended purpose, whilst not restricting the application of appropriate specified levels for therapeutic and diagnostic purposes.
在未对辐射用于治疗和诊断时的标准进行规定时,医疗器械的设计和制造应尽可能的避免患者、使用者和其他人员受到过多的辐射。
11.2 11.2.1 Intended radiation预期辐射
Where devices are designed to emit hazardous levels of radiation necessary for a specific medical purpose the benefit of which is considered to outweigh the risks inherent in the emission it must be possible for the user to control the emissions. Such devices shall be designed and manufactured to ensure reproducibilty and tolerance of relevant variable parameters.
当为特定医疗目的所设计的医疗器械,其释放的有害辐射物质的医疗成效大于其固有的风险时,使用者应尽可能的控制该辐射物质。这类医疗器械的设计和制造必须确保相关变化因素的再现性和所允许的偏差。
11.2.2 Where devices are intended to emit potentially hazardous, visible and/or
invisible radiation, they must be fitted, where practicable, with visual displays
and/or audible warnings of such emissions.
当医疗器械可能释放可见的和/或不可见的有害辐射时,可行时,医疗器械必须
安装辐射视频显示器和/或音响报警器。
11.3 11.3.1 Unintended radiation非预期辐射
Devices shall be designed and manufactured in such a way that exposure of patients, users and other persons to the emission of unintended, stray or scattered radiation is be reduced as far as possible.
医疗器械的设计和制造应尽可能的避免患者、使用者和其他人员受非预期辐射、杂散辐射或散射辐射的辐射。
Essential Requirements of the MDD (07/47/EC) 医疗器械指令(07/47/EC)基本要求Applicable
(Y/N)适用
(是/否)
Requirements
Standard 所符合的标
准要求
Manufacturers
Compliance/Records
制造商符合性文件/记录
Ok / Fail
符合/不符合
11.4 11.4.1 Instructions说明书
The operating instructions for devices emitting radiation must give detailed information as to the nature of the emitted radiation, means of protecting the patient and the user and on ways of avoiding misuse and of eliminating the risks inherent in installation.
释放辐射的医疗器械的操作说明书必须提供有关所释放的辐射的性质、保护患者和使用者的方法以及避免滥用的方法和消除安装风险的方法方面的详细内容。
11.5 11.5.1 Ionising radiation电离辐射
Devices intended to emit ionising radiation must be designed and manufactured in such a way as to ensure that, where practicable, the quantity, geometry and quality of radiation emitted can be varied and controlled taking into account the intended uses.
用于释放电离辐射的医疗器械的设计和制造,可行时,必须确保能够根据其预期用途对所释放的电离辐射的数量、几何结构和质量进行改变和控制。
11.5.2 Devices emitting ionising radiation intended for diagnostic radiology shall be
designed and manufactured in such a way, as to achieve appropriate image
and/or output quality for the intended medical purpose whilst minimising
radiation exposure of the patient and user.
释放电离辐射的医疗器械用于放射诊断时,其设计和制造应达到适当的图像和/
或特定医疗目的所需的输出质量,同时应减少患者和使用者所受到的辐射。11.5.3 Devices emitting ionising radiation intended for therapeutic radiology shall be
designed and manufactured in such a way as to enable reliable monitoring
and control of the delivered dose, the beam type and energy and where
appropriate the quality of the radiation.
释放电离辐射的医疗器械用于放射治疗时,应能够对辐射的输出剂量、光速类
型和能量以及一定的质量进行有效的监测和控制。
12.Requirements for medical devices connected to or equipped with an
energy source连接或装配有能源的医疗器械的要求
Essential Requirements of the MDD (07/47/EC) 医疗器械指令(07/47/EC)基本要求Applicable
(Y/N)适用
(是/否)
Requirements
Standard 所符合的标
准要求
Manufacturers
Compliance/Records
制造商符合性文件/记录
Ok / Fail
符合/不符合
12.1 Devices incorporating electronic programmable systems must be designed to
ensure the repeatability, reliability and performance of these systems
according to their intended use. In the event of a single fault condition (in the
system) appropriate means should be adopted to eliminate or reduce as far as
possible consequent risks.带有电子可编程系统的医疗器械的设计和制造必须根
据其预期用途确保这些系统的再现性、可靠性和性能。倘若(系统中)出现单
一故障,应尽可能采取措施消除或降低随之发生的风险。
12.1a For devices which incorporate software or which are medical software
in themselves, the software must be validated according to the state of
the art taking into account the principles of development lifecycle, risk
management, validation and verification.
对于装有软件或内置有医疗软件的医疗器械,软件必须在现有的研发生
命周期、风险管理、确认和验证规范下按照目前的工艺水平进行确认。
12.2 Devices where the safety of the patients depends on an internal power supply
must be equipped with a means of determining the state of the power supply.
当患者的安全取决于医疗器械的内部电源时,医疗器械的安装必须确保能够测
定电源的使用状态。
12.3 Devices where the safety of th e patient d epends on an external power supply
must include an alarm system to signal any power failure.
当患者的安全取决于医疗器械的外部电源时,医疗器械必须安装报警系统以便
对任何电力故障发出报警信号。
12.4 Devices intended to monitor one or more clinical parameters of a patient must
be equipped with appropriate alarm systems to alert the user of situations
which could lead to death or severe deterioration of the patient's state of
health.
用于监测患者的一个或多个临床参数的医疗器械必须安装适当的报警系统以提
醒使用者患者的健康状况严重恶化或可能会导致其死亡。
12.5 Devices must be designed and manufactured in such a way as to minimise the
risks of creating electromagnetic fields which could impair the operation of
other devices or equipment in the usual environment.
医疗器械的设计和制造应能够降低在寻常环境中使用时所产生的电磁场对其它
医疗器械或设备的操作造成损害的风险。
Essential Requirements of the MDD (07/47/EC) 医疗器械指令(07/47/EC)基本要求Applicable
(Y/N)适用
(是/否)
Requirements
Standard 所符合的标
准要求
Manufacturers
Compliance/Records
制造商符合性文件/记录
Ok / Fail
符合/不符合
12.6 Protection against electrical risks电气风险防护
Devices must be designed and manufactured in such a way as to avoid, as far
as possible, the risk of accidental electric shocks during normal use and in
single fault condition, provided that the devices are installed correctly.
医疗器械的设计和制造应尽可能避免在其安装正确的情况下,正常使用时或发
生单一故障情况时产生的意外电击风险。
12.7
12.7.1
Protection against mechanical and thermal risks机械风险和热量风险防护
Devices must be designed and manufactured in such a way as to protect the
patient and user against mechanical risks connected with, for example,
resistance, stability and moving parts.医疗器械的设计和制造应保护患者和使用
者免受例如阻力、稳定性和动件方面的机械风险。
12.7.2 Devices must be designed and manufactured in such a way as to reduce to
the lowest possible level the risks arising from vibration generated by the
devices, taking account of technical progress and of the means available for
limiting vibrations, particularly at source, unless the vibrations are part of the
specified performance.根据技术进步和可用于限制震动(特别是震源处的震
动)的方法,医疗器械的设计和制造应能够把医疗器械所产生的震动风险降低
到最低水平,除非震动是医疗器械本身所特有的性能。
12.7.3 Devices must be designed and manufactured in such a way as to reduce to
the lowest possible level the risks arising from the noise emitted, taking
account of technical progress and of the means available to reduce noise,
particularly at source, unless the noise emitted is part of the specified
performance.根据技术进步和可用于降低噪音(特别是噪音源处的噪音)的方
法,医疗器械的设计和制造应把其产生的噪音风险降低到最低水平,除非噪音
是医疗器械本身所特有的性能。
12.7.4 The terminals and connectors to the electricity, gas or hydraulic and pneumatic
energy supplies which the user has to handle must be designed and
constructed in such a way as to minimise all possible risks.供应给使用者操作
的与电流、天然气或液压能和气能相连的终端设备或连接器的设计和制造应能
够降低所有可能产生的风险。
Essential Requirements of the MDD (07/47/EC) 医疗器械指令(07/47/EC)基本要求Applicable
(Y/N)适用
(是/否)
Requirements
Standard 所符合的标
准要求
Manufacturers
Compliance/Records
制造商符合性文件/记录
Ok / Fail
符合/不符合
12.7.5 Accessible parts of devices (excluding any parts or areas intended to supply
heat or reach given temperatures) and their surroundings must not attain
potentially dangerous temperatures under normal use.在正常使用情况下,医
疗器械中易触及的部件(用于供应热量或达到指定温度的任何部件或区域除
外)及其周边的温度不能达到潜在的危险温度。
12.8 12.8.1 Protection against the risks posed to the patient by energy supplies or substances所供应的能量或物质对患者造成危害的风险的防护
Devices for supplying the patient with energy or substances must be designed and constructed in such a way that the flow rate can be set and maintained accurately enough to guarantee the safety of the patient and of the user. 为患者提供能量或物质的医疗器械的设计和制造应确保其流速可被设定并保持准确性,以充分保证患者和使用者的安全。
12.8.2 Devices must be fitted with the means of preventing and/or indicating any
inadequacies in the flow-rate which could pose a danger.
Devices must incorporate suitable means to prevent, as far as possible, the
accidental意外 release of dangerous levels of energy from an energy and/or
substance source.
医疗器械必须安装能预防和/或指出由于流速的不充分可能产生危险的设备。
医疗器械必须组装适当的设备以尽可能的预防来自于能源和/或物质源的能源意
外外泄的危险。
12.9 The function of the controls and indicators must be clearly specified on the
devices.
Where a device bears instructions required for its operation or indicates
operating or adjustment parameters by means of a visual system, such
information must be understandable to the user and, as appropriate, to the
patient.
控制键和指示灯的功能必须清晰地标注在医疗器械上。
当医疗器械有操作说明书或有通过目视系统指示其运行参数或平均参数的说明
书,这些说明书必须利于使用者理解,必要时还要利于患者理解。
https://www.sodocs.net/doc/b718679625.html,rmation supplied by the manufacturer制造商提供的资料
Essential Requirements of the MDD (07/47/EC) 医疗器械指令(07/47/EC)基本要求Applicable
(Y/N)适用
(是/否)
Requirements
Standard 所符合的标
准要求
Manufacturers
Compliance/Records
制造商符合性文件/记录
Ok / Fail
符合/不符合
13.1 Each device must be accompanied by the information needed to use it
safely and properly, taking account of the training and knowledge of
the potential users, and to identify the manufacturer考虑到每个潜在用
户的培训程度和知识水平,每台医疗器械必须附有如何安全和正确使用
该器械的资料,以及鉴定制造商的资料。
13.2 Where appropriate, this information should take the form of symbols. Any
symbol or identification colour used must conform to the harmonised
standards. In areas for which no standards exist, the symbols and colours
must be described in the documentation supplied with the device.当适用时,这
些资料应采用符号形式。所使用的任何符号或颜色鉴定法须符合协调标准。在
没有协调标准的地方,须在医疗器械所附带的文件中对符号和颜色进行描述。
13.3 The label must bear the following particulars:标签必须含有以下详细内容:
a) the name or trade name and address of the manufacturer. For
devices imported into the Community, in view of their distribution in
the Community, the label, or the outer packaging, or instructions for
use, shall contain in addition the name and address of the
authorised representative where the manufacturer does not have a
registered place of business in the Community;制造商的名称或贸易名
称和地址。当制造商在欧洲市场没有已注册的营业场所时,对于进口到欧洲
市场的医疗器械,考虑到其在欧洲市场销售,其标签或外部包装或使用说明
书还应包括授权代理人的姓名和地址;
b) the details strictly necessa ry to identify the device and the contents
of the packaging especially for the users;
使用者识别医疗器械及包装物品所须的详细资料;
c) where appropriate, the word "STERILE";
当适用时,在标签上标注“STERRILE”字体;
d) where appropriate, the batch code, preceded by the word "LOT", or the
serial number;当适用时,在“LOT”前标注上批号或者编号;
e) where appropriate, an indication of the date by which the device should be
used, in safety, expressed as the year and month;
当适用时,在标签上标注医疗器械的有效使用期,安全起见以年和月表示;
Essential Requirements of the MDD (07/47/EC) 医疗器械指令(07/47/EC)基本要求Applicable
(Y/N)适用
(是/否)
Requirements
Standard 所符合的标
准要求
Manufacturers
Compliance/Records
制造商符合性文件/记录
Ok / Fail
符合/不符合
f) where appropriate, an indication that the device is for single use. A
manufacturer's indication of single use must be consistent across
the Community;当适用时,注明医疗器械做一次性使用。制造商的一次性
使用标识在欧洲市场内必须一致;
13.3 g) if the device is custom made, the words "custom made device";
如果医疗器械是定制的,应标注“定制的医疗器械”;
h) if the device is intended for clinical investigation s, the words "exclusively for
clinical investigations";
如果医疗器械用于临床研究,应标注“专供临床研究之用”;
i) any special storage and/or handling conditions;
任何特殊的存储和/或处理条件;
j) any special operating instructions;任何特殊的操作说明书;
k) any warnings and/or precautions to take;
任何警告和/或采取的预防措施;
l) year of manufacture of active devices other than除了 those covered by e).
This indication may be included in the batch or serial number;
有源医疗器械除了满足e)中所列的信息,还应标注上制造年份。制造年份
可能包含在批号或编号中;
m) where applicable, method of sterilisation.当适用时,标注上灭菌方法。
13.4 If the intended purpose of the device is not obvious to the user, the
manufacturer must clearly state it on the label and in the instructions for use.
如果医疗器械的用途不太明显,制造商应在使用说明书和标签上陈述清楚。13.5 Wherever reasonable and practicable, the devices and detachable
components must be identified, where appropriate in terms of batches, to
allow all appropriate action to detect any potential risk posed by the devices
and detachable components.当合理和可行时,当适用时必须根据批号对医疗器
械及其部件进行识别,以便采取所有适当措施查明由医疗器械及其部件引起的
任何潜在风险。
13.6 Where appropriate, the instructions for use must contain the following
particulars:当适用时,使用说明书必须含有以下详细资料:
Essential Requirements of the MDD (07/47/EC) 医疗器械指令(07/47/EC)基本要求Applicable
(Y/N)适用
(是/否)
Requirements
Standard 所符合的标
准要求
Manufacturers
Compliance/Records
制造商符合性文件/记录
Ok / Fail
符合/不符合
a) the details r eferred to in 13.3, with the exception of d) and e);
第13.3中所述的除 (d)和(e)之外的内容;
b) the performances referred to in section 3 a nd any undesirable side effects;
条款3中提到的性能和任何不良副作用;
c) if the device must be installed with or connected to other medical devices
or equipment in order to operate as required for its intended purpose,
sufficien t details of it s characteristics to identify the correct devices or
equipment to use in order to obtain a safe combination;如果器械必须同其他医疗器械或设备安装或连接在一起,以便按其预定用途要求进行工作,爲了实现安全的组合,要标明识别使用器械和设备的特性的详细细节。
d) all the information needed to verify whether the device is properly installed
and can operate correctly and safely, plus details of the nature and
frequency of the maintenance and calibration needed to ensure that the devices operate properly and safely at all times;验证医疗器械是否正确安装和能否正常运行和安全运行所需的所有资料,以及确保医疗器械一直正确运行和安全运行所需的维护和校准的性质和频率方面的详细资料;
e) where appropriate, information to avoid certain risks in connection with
implantation of the device;当适用时,预防与医疗器械的植入有关的必然风险方面的资料。
f) information regarding the risks of reciprocal interference posed by the
presence of the device during spe cific investigations or tr eatment;在进行特殊研究或治疗时由于医疗器械的存在而产生的相互干扰风险方面的资料。
g) the necessary instructions in the event of damage to the sterile packaging
and, where appropriate, details of appropriate methods of re-sterilisation;
无菌包装遭到损坏时的必要说明,当适用时,还应包括适当的再灭菌方法方面的详细内容;
手机结构设计checklist
手机结构设计检查表一.通用性项目 二.功能性项目 1.镜片Sub Len s 镜片的工艺(IMD/IML/模切/注塑+硬化/电铸+模切)
镜片的厚度及最小厚度 IMD/IML/注塑镜片P/L,draft,radius? 固定方式及定位方式,最小粘接宽度是否大于1.5mm? 窗口(VA&AA)位置是否正确 镜片本身及固定区域有无导致ESD问题的孔洞存在 周边的电铸或金属件如何避免ESD 小镜片周边的金属是否会对天线有影响(开盖时) 2.转轴Hing e 转轴的直径 转轴的扭力 打开角度(SPEC) 有无预压角度(开盖预压为4-6度,建议5度 装拆有无空间问题? 固定转轴的壁厚是多少,材料(推荐PC GE C1200HF或者三星HF1023IM) 转轴配合处的尺寸及公差是否按照转轴SPEC? 3.连接FLIP(SLIDE)/BASE的FPC 1) FPC的材料,层数,总厚度 2) PIN数,PIN宽PIN距 3)最外面的线到FPC边的距离是多少(推荐0.3mm) 4) FPC内拐角处最小圆角要求大于1mm,且内拐角有0.20mm宽的布铜,防止折裂. 5)有无屏蔽层和接地或者是刷银浆? 6) FPC的弯折高度是多少(仅限于SLIDE类型) 7) FPC与壳体的长度是否合适,有无MOCKUP 验证 8)壳体在FPC通过的地方是否有圆角?多少?推荐大于0.20mm. 9) FPC与壳体间隙最小值?(推荐值为0.5mm) 10) FPC不在转轴内的部分是否有定位及固定措施? 11)对应的连接器的固定方式 12) FPC和连接器的焊接有无定位要求?定位孔? 13)补强板材料,厚度 4.LCD 模组 主副LCD的尺寸是否正确及最大厚度 主副LCD的VA/AA区是否正确 主副LCD视角,6点钟还是12点钟? 副LCD是黑白/OLED/CSTN/TFT?相应的背光是什么? 副板是用FPC还PCB? PCB/FPC的厚度及层数. LCD模组是由供应商整体提供吗? 如果不是,主LCD如何与PCB/FPC连接?连接器类型及高度or HOTBAR? 副LCD如何与PCB/FPC连接?连接器类型及高度or HOTBAR? FPC/PCB上有无接地?周边有无露铜 有无SHIELDING屏蔽?厚度,材料,如何接地? 元件的PLACEMENT图是否确定? 有无干涉? 主副LCD的定位及固定 LCD模组的定位及固定 LCD模组有无CAMERA模组,是否屏蔽?
新项目Checklist
New Product Development Tollgate Checklist Feasibility For Quote (stageⅠ)可行性阶段一Proto Type(stage Ⅱ)打样(阶段二) Pilot Run (stage Ⅲ)试生产(阶段三)Mass(1 Year review)(stage Ⅳ)量产(阶段四)
Continue to Next Stage继续下阶段Another Review Required and Date需重新review及时间Terminate the Project终止该项目Notice to Client通知客户 New Product Requirement Checklist Details新产品评审要求细则 #1- Drawing / Print:Need to confirm the latest revision level drawings available. Require all assembly, sub-assembly and individual component drawings are provided from customer. 图纸:需要确认并获得最新版本的图纸,要求客户提供所有组件,分部组件和子件的图纸 #2- Critical Material: Need to identify material requirements for the project to confirm availability in China and available material substitutions; this should include any supplied material as well as the manufacturer (such as steel mill, paint manufacturer or OSP) of this material. 关键材料:需要识别该项目的材料要求并确认在国内可购买以及有可用的替代材料,此要求除了制造商如钢厂,涂料厂商或外协厂商外,还需包含其它原材料及物料 #3- Customer Standards & Specifications:Acquire all customer standards, specifications and requirements for the part numbers being considered for quoting purposes. Ask for the following customer documents: Supplier Quality Manual, Specifications listed on drawing / print, PPAP, Material Certificate Requirements or any third party test requirements. 客户标准及规格:对正在评估报价的项目料号取得客户标准,规范和要求。获得客户相关文件:如供应商质量手册,列在图纸上的规范清单,PPAP,材料认证要求或任何第三方测试要求
项目清单(project charter checklist)
项目清单(project charter checklist) 又名:任务说明检查单(mission statement checklist) 概述 项目清单用于确保整个小组明白该做的事情以及采取的方式。 适用场合 ·新成立的小组召开第一次或笫二次会议时; ·当小组获得新的或者修改过的任务时。 实施步骤 讨论下面的问题: 1是否清楚管理者对我们的期望? 2是否清楚为什么该项目对组织来说是重要的?是否清楚该项目对组织的总体目标的切入点在哪里? 3我们的项目是否仅是一个较大质量改进过程的一部分?在这个过程中,我们的项目处在什么位置?以及它应该从哪儿开始和到哪儿结束? 4项目范围的界定是否清楚?什么将超出我们的权限范围? 5目标是否现实? 6该项目是否可行?该任务是否与我们对过程或系统的了解相吻合? 7在部门内、外部,我们都需要哪些资源? 8我们小组是否有合适的人员去完成该任务? 9在我们小组以外,哪些人将对我们的工作至关重要? 10谁将支持我们?准将反对我们?谁将中立?我们怎样才能接触到这些人? 11怎样表达反对意见?我们如何确保让所有的反对意见都表达出来? 12我们将通过什么方式判断我们已经成功?其他人将通过什么方式判断我们是否已经成功? 示例 技术支持部门的一个小组被授予这样一个任务:“通过对过程步骤进行考察,改善报表准备。”该小组利用项目清单论述了各项工作,问题就变得很清楚了。这是他们讨论的一些问题:1经理所期望的提高程度是多少?他们感兴趣的改善种类是什么?应该对报表准备的所有方面给予同等的关注吗? 2报表准备是如何影响组织工作的?哪些方面正给我们或者我们的顾客带来麻烦? 3我们的工作应该致力于哪些过程?我们所涉及的只有书面报表阶段还是形成报表的整个调查过程? 4我们是否被允许到所凋查的部门内部研究导致影响报表问题的症结所在?管理者对于报表的评价是什么? 5管理者希望这个项目持续多长时间?考虑到我们的其他职责,这个时间是否足够? 6这个项目能否起作用取决于对我们某些问题的回答。 7管理层是否通知了客户部的经理:我们需要他们的员工抽出一些时间为我们提供信息?他们是否也通知了我们部门,特别是秘书?认为我们的工作拥有了合适的人选。 8我们认为拥有了合适的人选。 9关键人员:我们部门其他的报表人员——秘书以及经理。 10报表的撰写者不会去追求以不同的方式工作。如果我们可以找到减少秘书工作量的方法,就会得到他们的支持。 11要确保考虑并解决全部的报表人员提出的问题。当我们提出改革时,应该对他们进行排除自身利益的培训。 12我们如何判断我们已经获得成功?经理用何种方式判断?因为我们不清楚他们所期望的
汽车内外饰工艺数据checklist
仪表板内饰工艺数据checklist 1、是否根据确定方案进行设计; 2、数模分层符合公司标准; 3、零部件编号是否符合标准; 4、电子文档命名、版本编制是否符合规范; 5、零部件坐标系的统一性(模型一律采用整车坐标系); 6、检查**件与点云偏差,车身结构件数模有安全配合是否的面与测量云的偏差± 0.3;自由曲面数模与测量云偏差±1;安装孔位与测量点云的偏差± 0.5; 7、明细表中件号、数模是否对应; 8、零件成型方法是否合格; 9、仪表板最高的及两端点,校核仪表板位置参数,是否满足人机工程要求; 10、各零件的成型工艺是否确定(如注射、挤出、模压、压延、铸型、吹塑等成型的方法); 11、脱模方向是否正确; 12、检查塑料零件壁厚是否均匀一致,壁厚不均匀处易产生气泡和收缩变形,甚至产生断裂; 13、检查数模内部是否有凹陷(即复角部分),凹陷存在不便出模; 14、选用合适的脱模斜度和适当的脱模剂,脱模斜度大小与塑料件材料的性质、厚度、形状等有关;
15、载塑料零件上,是否避免锐角及直角过渡; 16、安装方式是否正确; 17、正确的选择定位尺寸基准,应尽可能使设计基准和工艺基准重合,避免装配过程中,误差的积累过大; 18、经常所装的零部件,为了更换方便,应以螺栓成自攻螺钉和簧片螺母配合紧固连接; 19、明确安装工具,预留所需的被动空间; 20、在安装过程中,需要进行装配调整的零部件要考虑孔位的合理布置及适当地预留间隙调整; 21、考虑到仪表板内线束的固定,明确线卡固定点及固定方式,钣金上的线束的过孔是否加以保护套成翻边结构; 22、检查保险杆外表面在X方向是否有负面保证模具成型后外表面的完整和美观; 23、检查外表面面与面的偏差是否超标; 24、检查外表面可增厚性; 25、检查零件的强度是否适当,是否有强度薄弱的区域(薄弱的区域需增加加强筋); 26、检查零件的材料选用是否适当(如毛面需要镀铬地零件应选用同ABS 等可镀铬材料,不能选用PP难镀的材料; 27、在塑料件结构设计中,为避免转角处应刀集中,应采用圆弧过渡,这对于模具制造及使用寿命足很有利的; 28、对于保险杆要进行相关国家法规的检查(接近角、离车角等);
产品结构设计等方面的checklist
模具的checklist表: 产品名称模具编号材料收缩率 序号内容自检确认 1与客户交流清楚外观面位置及外观要求如镜面,皮纹,亚光等。 2清楚产品的安装方向,产品的出模方向及它们之间的关系。 3产品在出模方向无不合理结构。 4壁厚合理,壁厚均匀,没有过薄,过厚及壁厚突变。 5圆角齐全,所有外观面倒圆角(特殊要求除外),所有非外观面倒圆角,非外观面圆角足够大。且圆角处壁厚均匀,无漏掉的圆角。 6脱模斜度齐全,正确,无放反的情况,脱模斜度足够大,已用DRAFTCHECK命令进行检查。7透明件,皮纹处理的外观面,插穿面脱模斜度足够大,满足标准。 8透明件已考虑外观效果,可见结构,并与客户进行交流。 9需贴膜的件已经考虑到膜在实际安装方向的定位, 10电镀件装配考虑到镀层厚度和装配间隙, 11一面用插接,一面用卡爪的结构已考虑到装配过程中是否有与外观干涉,是否有造成外观面破坏的情况,卡爪是否易断 12加强筋高度,宽度,脱模斜度结构及工艺均合理。 13外观件检查产品结构如壁厚,加强筋(尤其是横在制品侧壁的筋考虑与侧壁的防缩)、螺钉柱等不会引起缩水,已采取防缩措施。 14产品变形,收缩等注塑缺陷轻微,且已与客户协商,得到客户的书面认可。 15需出斜顶,滑块,抽芯的结构活动距离及空间足够,结构能否简化。 16产品无引起模具壁薄,尖角等不合理结构。 17带嵌件的产品考虑嵌件在模具中的牢固固定,内桶底的嵌件要求将嵌件和包嵌件的胶位合并到一起作为模具嵌件。 18与客户交流清楚分型面的位置,外观面滑块,抽芯允许的夹线位置。 19备份产品已检查所有修模报告及更改记录并进行了更改,重要装配尺寸进行了样件的实际测绘验证。 笔记本的CHECKLIST DesignCheckListBySub-Assy. 1.U-Case 1-1上下盖嵌合部份 1-1-1上下盖PL是否Match 1-1-2Lip是否完成,是否符合外观要求(修饰沟) 1-1-3侧壁之TAPER/与下盖是否配合/考虑到开模 1-1-4上下盖之配合卡勾共几处,是否位置match 1-1-5卡勾嵌合深度多少 1-1-6卡勾两侧有无夹持Rib,拆拔时是否易断裂 1-1-7卡勾是否造成侧壁缩水(如果太厚) 1-1-8公模内面形状(如各处高度). 1-1-10PL切口处是否有刀口产生(全周Check) 1-2BOSS 1-2-1上下盖BOSS孔位是否相合 1-2-2BOSS尺寸是否标准化,内缘有没有倒角
新业务项目业务案例checklist
新业务项目业务案例检查表 新业务项目在进行“业务案例”分析之前,需要明确如下问题。这些问题已被明确的程度,以及其被明确的可能性是新业务项目是否可以作为“业务案例”研究的关键。 一、市场分析 o该新业务满足了哪些市场需求? o该新业务平台将推出哪些应用服务? o该新业务的政策、行业标准、行业规定因素 o该新业务各应用的目标客户群定义 o该些目标客户群的规模以及发展趋势? o市场渗透的策略以及市场开拓的预期(市场份额发展趋势等) o客户使用各应用服务的频率,即服务使用量发展预测 二、业务分析 o各应用服务的完整业务流程描述 o该服务各应用的业务模式(上下游合作伙伴、合作方式) o该新业务各应用实现的技术方案 o该新业务提供的网络建设方案 o该新业务各应用服务推出计划(何时开始提供哪些服务) o该业务的生命周期是否有限?还是可以视为永久业务?(是否考虑项目终值) 三、收益分析(基础情景) o各应用服务的收入模型:定价方式、费率标准以及发展趋势 o各应用服务的收入预测 o各应用服务收入发展规律判断 o其它可量化收益 o其它不可量化收益 o收益发展规律 四、投资分析(基础情景) o项目预可研、可研成本 o试运行阶段的投资(前期勘测、设计费用、建安成本、网络投资、运营成本) o实现该新业务的各项应用需要对现有网络进行哪些建设、改造? o设备、硬件软件系统选型方案或参考方案及造价成本 o系统集成成本 o建安成本 o该些改造的投资规模、计划及资金来源? o该些网络投资的扩容与升级计划? o该些扩容、升级的投资规模、计划及资金来源? o其它需要资本化到该新业务上的投资规模、计划及资金来源? o各项投资的资金支付计划 o投资发展规律 五、运营成本分析(基础情景) o各应用服务提供与合作伙伴间的结算原则及成本 o网络运营维护支持计划及边际网络运营维护成本 o传输计划以及边际传输成本
华为单板硬件设计审查评审表checklist
单板硬件设计审查评审表 文档编号:文档名称: 文档作者:文档完成时间:项目经理: 所属单板名称: 1、可读性评价: □很好□较好□一般□较差 说明:文档是否表达清晰,逻辑条理分明,表达形式通用,使具有一定技术背景的工程师容易读懂。如:在难懂的地方增加注释,在适当时采用图文并茂的方式等。选择认可的项打叉或打勾。 2、准确性评价: □很好□较好□一般□较差 说明:指文档是否对其中的技术内容能表达准确,对其中设计的测试方法有其操作性,并且准确有实效,不应该有关键技术表达错误等。选择认可的项打叉或打勾。 3、规范性评价: □很好□较好□一般□较差 说明:指文档的内容和形式是否是规范的,如:文档是否按模板来写;在特殊的情况下不使用模板而写的文档其封面格式、字体、主要内容顺序是否和相应的文档模板类型的要求是否一致等。选择认可的项打叉或打勾。 4、完备性评价 完备性总评: □很好□较好□一般□较差 说明:指文档包含的测试项目是否完整(即:没有漏测现象等),本次测试总体上对测试指导书的遵从程度和测试深度。可对照附录的内容进行判断。 总评: 说明:概括总结该文档的优点、缺点及改进建议 评审人签字:评审日期:联系电话: 附单板设计审查项目列表:
请参照此表,审查过的项目请打(9),未审查的项目请打(x),单板无此审查项目可不填。 1.单元电路审查: 1.1滤波电路审查 1.审查电路中有无设计电源滤波器。有无审查() 2.审查电路中电源滤波器的形式是否有效,是否为单电 容型或单电感型,而未采用П形电源滤波器。有无审查() 3.对单板的П形电源滤波器参数进行审查。有无审查() 1.2ID电路审查 1.审查ID电路的形式是否符合规范电路的要求。有无审查() 2.审查ID电路的参数是否正确。有无审查() 3.审查ID电路是否有隔离电阻或隔离芯片。有无审查() 4.在沿用未能提供正确ID处理的旧母板时,单板是 否进行相应的处理。有无审查() 1.3主备倒换电路审查 1.审查主备倒换电路是否为主倒备型电路。有无审查() 2.主备电路设计中是否考虑到单板复位后一段时间 内该板一直设为备用,以更有效防止备抢主。有无审查() 3.电路中是否考虑在主板复位时,自动转为备板,两块 板同时复位时,自动将0号板设为主用,1号板设为备用。有无审查() 4.在备板插拔时,由于插针接触或脱离的次序先后 有别,会否导致备抢主现象。有无审查() 5.备板在插入的过程中,会否有可能导致主板的状态不正常。有无审查() 6.是否未将/Reset信号引入主备倒换电路,可否存在隐患。有无审查() 7.主备倒换电路能否在单板所有的故障状态下均 能进行正常的倒换,包括主板通讯中断时的自动倒 换,CPU故障时的自动倒换等情况。有无审查() 8.主备倒换电路与系统的时序配合能否满足系统实时倒换的要求。有无审查() 9.若单板有一一对应关系,有否考虑到相关单板的联动倒换。有无审查() 10.设计中是否考虑到本板通过光纤,双绞线输入的重要信号丢失 时的自动倒换.有无审查() 1.4复位、WDT电路审查 1.硬件设计中不推荐使用可关闭的WDT系统,即计数器清零电路应
代码大全核心checklist
为了更好的评估代码写的哪里有问题,我把《代码大全》里核心的部分checklist整理出来了,大家可以大概过一遍,不一定每写完一个程序都要一条一条的去检查,但心里应该有这么一 张检查表,在写代码和review代码时自然而然的想起来。 设计 设计是否经过多次迭代,并最终决定了最好的一个? 是否同时使用自上而下和自下而上的方法来解决设计问题? 类与类之间的交互关系是否已经设计为最小化? 设计被划分为层次吗? 你对把这一程序分解成为子程序,包和类的方式感到满意吗? 程序是不是易于维护? 设计是否精简?设计出来的每一个部分都绝对必要吗? 整体而言,你的设计是否有助于最小化偶然性和本质性的复杂度吗? 类的设计 你是否把程序中的类都看做是抽象数据类型了?是否从这个角度评估它们的接口了? 类是否有一个中心目的? 类的命名是否恰当?其名字是否表达了其中新目的? 类的接口是否展现了一致的抽象? 类的接口是否能让人清楚明白的知道如何用它? 类的接口是否抽象,使你能不必顾虑他是如何实现其服务的?你能把类看做黑盒子吗? 类提供的服务是否足够完整,让其它类无需动用其内部数据? 是否已从类中去除无关信息? 是否考虑过把类进一步分解? 在修改类时是否维持了其接口的完整性? 是否把成员的可访问性降到最小? 是否避免暴露类的数据成员? 类是否避免对其使用者,包括其派生类会如何使用它做了假设? 类是否不依赖于其它类?它是松散耦合吗? 继承是否只用来建立一个is a关系?派生类是否遵循了LSP原则。 继承层次是否很浅? 类中是否只有大约七个或者更少的成员? 是否把类直接或者间接调用其他类的子程序的数量减到最少? 类是否在绝对必要时才与其他类写作? 是否在构造函数中初始化了所有的数据成员? 子程序 创建子程序的理由充分吗? 一个子程序中所有适合单独提出的部分是不是已经被提出到单独的子程序中了?
投标checklist-新版.doc
序号投标步骤确认check点确认结果时间(天) 项目类型、规模是□否□ 接到投标任 务,与客户经 投标周期是□否□ 理沟通了解 项目细节,获 取标书 明确项目组人员配置状况是□否□0.5 根据项目情况,计划好会议室大约预定是□否□ 联系秘书,预 定投标作战周期并预定 室 阅读标书是□否□ 阅读标书,分配投标任务提取标书关键信息是□否□ 划分投标任务是□否□ 制定总体投标计划是□否□ 0.5 输出投标分工表是□否□ 召集并组织开工会是□否□ 分配投标任务,指定答标责任人是□否□ 与项目组成员澄清任务项中是否有缺是□否□ 失,任务分配是否正确 开工会投标项目计划及时间点的澄清是□否□ 评审并确定标书的框架结构是□否□ 0.5 确定投标模式及投标主体是□否□ 标书中关于封装、打印件数、电子件要是□否□ 求、交标时间、地点等细节信息。要和 项目组成员一一过一下 准备盖章件、提交印章申请流程是□否□ (渠道投标)在PRM 提交制造商授权函 是□否□ 与售后服务承诺函业务审批 启动投标项目申请流程(直投直签)填写中国区销售业务审批 电子流,在PRM 中启动招标书客户化合 是□否□ 0.5 同文本评审、售后服务承诺函评审、提 交投标保证金申请、对外付款电子流等 其他投标相关流程申请是□否□ 商务答标文件准备是□否□ 答标文件准 备技术答标文件准备是□否□ 服务答标文件准备是□否□ 审核投资人资质文件是否符合标书要求是□否□ 3 (渠道项目重点关注代理商的资质) 商务答标文件初稿评审(第一轮)是□否□ 商务答标文件初稿评审(第二轮)『可选』是□否□ 标书评审技术答标文件初稿评审(第一轮)是□否□技术答标文件初稿评审(第二轮)是□否□
嵌入式软件可靠性设计规范checklist
嵌入式软件可靠性设计规范汇总
43.高级报警显示:红色,1.4Hz~ 2.8Hz,信占比率20%~60%开 44.中级报警显示:黄色,0.4Hz~0.8Hz,信占比率20%~60%开 45.低级报警显示:蓝绿色或者黄色,常开,信占比率100% 46. 高优先级和中优先级的报警上、下限设置值,一旦超出可能引起较严重后果的非合理报警数值区域时,均需加单独的对话弹出框予以提醒操作者 47. 默认的报警预置不允许修改,并提供让用户能恢复到出厂默认报警设置的操作途径 48.做报警日志记录,为以后的故障分析、维修检查或商业纠纷提供依据 与硬件接口的软件49. 数据传输接口的硬件性能限制了数据传输速率的提高,在确定波特率前,要确认硬件所能承受的最高传输率,光耦、485、232、CAN、传输线上有防护 器件(TVS或压敏电阻)的端口 50.硬件端口读进来的数据必须加值域范围的判断 51.硬件端口读取数据,必须加可控时间或次数的有限次限制 52.A/D的位数比前端放大电路的精度要求略高即可,并通过数学计算验证 53. 对运动部件的控制,正向运动突然转向反向运动时,必须控制先正向减速到0,然后再反向加速的控制方式 54. 运动部件停机后、再快速启动的工作控制方式是不允许的。须停机、开机、delay延时、再启动执行机构,以确保执行机构先释放原来运动状态的惯性,然后再从静态下启动 55. 运动部件都有过渡过程特性,软件驱动时的上升沿和下降沿的过渡特性会 直接影响到硬件的安全和执行效果 56. 板卡启动时,先initMCU、然后Delay、然后initIO,以确保各芯片的上电 电源都已经稳定下来再启动工作 57. 对采集自有可能受到干扰的模拟端口输入的数字量数据,一定要加上、下 限、Δ/Δt、规律性干扰的滤波措施三个方面的容错性机制 58. 对数字端口传输数据可以连续传输两遍,以防范随机性偶发干扰,实时性要求较高的,可以连续传三遍,2:1判定 59. 模块之间的数据通信联络,用周期性读取的方式、或请求-应答的方式传送 数据,一旦超出周期性时间要求,或未应答,则判定硬件失效,需有软件的
ICF checklist
ICF CHECKLIST Version 2.1a, Clinician Form for International Classification of Functioning, Disability and Health This is a checklist of major categories of the International Classification of Functioning, Disability and Health (ICF) of the World Health Organization . The ICF Checklist is a practical tool to elicit and record information on the functioning and disability of an individual. This information can be summarized for case records (for example, in clinical practice or social work). The checklist should be used along with the ICF or ICF Pocket version. H 1. When completing this checklist, use all information available. Please check those used: [1] written records [2] primary respondent[3] other informants [4] direct observation If medical and diagnostic information is not available it is suggested to complete appendix 1: Brief Health Information (p 9-10) which can be completed by the respondent. H 2. Date __ __ /__ __/ __ __ H 3. Case ID _ _ , __ __ __ , __ H 4. Participant No. __ __ , __ __ , __ __ __ Day Month Year CE or CS Case No. 1st or 2nd Evalu FTC Site Participant A.DEMOGRAPHIC INFORMATION A.1 NAME (optional)First ____________________ FAMILY_______________________ A.2 SEX (1) [ ] Female(2) [ ] Male A.3 DATE OF BIRTH _ _/_ _/_ _(date/month/year) A.4 ADDRESS (optional) A.5 YEARS OF FORMAL EDUCATION _ _ A.6CURRENT MARITAL STATUS: (Check only one that is most applicable) (1) Never married[ ] (4) Divorced[ ] (2) Currently Married[ ] (5) Widowed[ ] (3) Separated[ ] (6) Cohabiting[ ] A.7 CURRENT OCCUPATION(Select the single best option) (1) Paid employment [ ] (6) Retired[ ] (2) Self-employed [ ](7) Unemployed (health reason)[ ] (3) Non-paid work, such as volunteer/charity[ ] (8) Unemployed (other reason)[ ] (4) Student[ ](9) Other[ ] (5) Keeping house/House-maker[ ](please specify) ____________ A.8 MEDICAL DIAGNOSIS of existing Main Health Conditions, if possible give ICD Codes. 1.No Medical Condition exists 2.…………………….. ICD code: __. __. __.__. __ 3.…………………….. ICD code: __. __. __.__. __ 4.…………………….. ICD code: __. __. __.__. __ 5. A Health Condition (disease, disorder, injury ) exists, however its nature or diagnosis is not known ICF Checklist? World Health Organization, September 2003.Page 1
性能测试checklist
如果有朋友想到更多的检查项,也希望可以留言大家一起讨论 1. 开发人员是否提交了测试申请? 2. 测试对象是否已经明确? 3. 测试范围是否已经明确? 4. 本次不被测试的范围是否已经明确? 5. 测试目标是否已经明确? 6. 何时开始性能测试? 7. 何时终止一轮性能测试? 8. 性能测试需要做几轮? 9. 所需的测试环境是什么?是否已经到位并配置完成?(包括硬件、软件、网络等10. 所需的测试工具是什么?是否已经到位并保证可以正常使用? 11. 被测系统的版本是否已经明确?是否已发布?从哪里可以获得?是否已经部署成功? 12. 被测系统的相关功能是否已经正确实现? 13. 压力点是否已经明确?响应时间的计算方式是否已经明确? 14. 本次测试工作参考的文档有哪些? 15. 场景是否已经设计完成并记录在场景管理文档中? 16. 每个场景是否有明确的测试意图、前置条件和详细的设置? 17. 脚本是否已经录制并调试通过? 18. 是否已经明确了哪些地方需要参数化?
19. 是否已经明确了各个参数的取值方式? 20. 是否已经为参数化的部分准备了必须的数据? 21. 是否已经准备了相应历史数据量? 22. 是否已经准备了相应的数据恢复方法?(例如准备一个SQL 语句用来恢复数据环境 23. 在Controller 中对多VU 、多次迭代的情况是否已经调试通过? 24. 在Controller 中Result 的路径设置是否正确? 25. 在Controller 中检查脚本选择是否正确? 26. 在Controller 中检查VU 数量设置是否正确? 27. 在Controller 中检查集合点是否禁用/启用? 28. 在Controller 中检查VU 加载策略是否设置正确? 29. 在Controller 中检查迭代次数是否设置正确? 30. 在Controller 中检查迭代间隔设置是否正确? 31. 在Controller 中检查日志是否禁用/启用? 32. 在Controller 中检查Think_Time 是否回放? 33. 在Controller中检查是否为UNIX服务器和Load Generator机添加了资源监视器并确认可以收到性能数据? 34. 在Controller 中检查是否为其他必要的资源添加了资源监视器,并确认可以收到性能数据(例如Oracle , WebSphere ? 35. 在Controller中检查Load Generator机是否可以连上? 36. 检查场景管理文档中是否添加了新的“场景执行情况”并, 记录了运行
PCB-Checklist
PCB-Checklist
阶段项 目 序 号 检查内容EDA 设计 EDA 复审 EDA 确认 备注 资料输入阶段1.在流程上接收到的资料是否齐全(包括:原理图、*.brd文件、料单、PCB设计说明以及PCB 设计或更改要求、标准化要求说明、工艺设计说明文件) 2.3确认PCB模板是最新的 3.确认模板的定位器件位置无误 4.PCB设计说明以及PCB设计或更改要求、标准化要求说明是否明确 5.4确认外形图上的禁止布放器件和布线区已在PCB模板上体现 6.比较外形图,确认PCB所标注尺寸及公差无误, 金属化孔和非金属化孔定义准确 7.5 确认PCB模板准确无误后最好锁定该结构文件,以免误操作被移动位置 布局后检查 阶段器 件 检 查 8.确认所有器件封装是否与公司统一库一致,是否已更新封装库(用viewlog检查运行结果)如 果不一致,一定要Update Symbols 9.母板与子板,单板与背板,确认信号对应,位置对应,连接器方向及丝印标识正确,且子板有 防误插措施,子板与母板上的器件不应产生干涉 10.元器件是否100% 放置 11.打开器件TOP和BOTTOM层的place-bound,查看重叠引起的DRC是否允许 12.M ark点是否足够且必要 13.较重的元器件,应该布放在靠近PCB支撑点或支撑边的地方,以减少PCB的翘曲 14.与结构相关的器件布好局后最好锁住,防止误操作移动位置 15.压接插座周围5mm范围内,正面不允许有高度超过压接插座高度的元件,背面不允许有元件或 焊点 16.确认器件布局是否满足工艺性要求(重点关注BGA、PLCC、贴片插座) 17.金属壳体的元器件,特别注意不要与其它元器件相碰,要留有足够的空间位置 18.接口相关的器件尽量靠近接口放置,背板总线驱动器尽量靠近背板连接器放置 2
检查单(checklist)
检查单(checklist) 又名:工作程序(work procedure),实证检查单(confirmation check sheet) ?概述 检查单是对需要考虑或完成的条目、行动和问题进行核对之前,预先确定的清单列表。检查单是检查表的一种,是门类工具。 ?适用场合 ·当一个有很多步骤的过程或程序必须反复进行时; ·当一个过程或程序由于进行得不频繁而可能被遗忘时; ·当要开始一个有很多步骤或细节的活动,尤其是以前没做过时; ·当开始一个将要持续很长时间的项目时; ·当一组问题和事项可以用在不同的情景时; ·当一个过程的行动或步骤必须按照某个顺序进行时; ·当不遗漏列表上的条目变得非常重要时。 经常使用检查单的特殊情形: ·工作程序 ·安全检查 ·审计 ·项目复审 ·竞赛筹划 ·会议筹划 ?实施步骤 如果检查单不存在 1确定检查单的目的和范围。 2进行研究或者用头脑风暴法确定在清单上需要列出的条目。 3写下这些条目,尽可能清楚、简洁。 4确定条目的顺序是否重要。如果重要,安排合适的顺序。同样,确定如果一个步骤被遗漏所需的弥补措施。 5如果清单需要被反复利用,使用表格填写清单条目,并留出位置填写核对信息或完成条目的时间,如果步骤的顺序非常重要,确保清单中包含说明信息以及一个步骤被遗漏时的弥补措施。 6如果清单被反复利用,与没有帮助制作清单的人们一起检查它。寻找错误、遗漏、不合理的顺序或者不清楚的语言。 如果已有检查单,或者已经自己准备好了清单 7在过程、程序或行动进行的过程中,保持检查单处于容易得到的位置。 8每次执行过程、程序或行动的时候参阅一下检查单。 9在每个条目完成时,核对并填写完成日期。 10当检查单填满时,保证每个条目都经过核对。 11如果过程或程序的步骤排序非常重要,或者前一个条目没有检查之前后一个条目已经做完,则参阅检查单中处理遗漏步骤的说明。 ?示例 本书中质量改进过程的10个步骤,或者一个组织所进行的流程都是一个检查单。书中每一套程序也都是一个检查单。书中已有的检查单包括项目章程清单和5W2H。日常生活中常见的检查单包括会议日程安排、要做的事情列表、购物单和玩具装配说明书。作为在质量改进过程中使用检查单的例子,请参阅第4章圣鲁克医院和ZZ-400案例。 ?注意事项
原理图checklist
原理图checklist
原理图checklist 类别描述 检视规则原理图需要进行检视,提交集体检视是需要完成自检,确保没有低级问题。 检视规则原理图要和公司团队和可以邀请的专家一起进行检视。 检视规则第一次原理图发出进行集体检视后所有的修改点都需要进行记录。检视规则正式版本的原理图在投板前需要经过经理的审判。 差分网络原理图中差分线的网络,芯片管脚处的P和N与网络命令的P和N 应该一一对应。 单网络原理图中所有单网络需要做一一确认。空网络原理图中所有空网络需要做一一确认。 网格1、原理图绘制中要确认网格设置是否一致。 2、原理图中没有网格最小值设置不一致造成网络未连接的情况。 网络属性确认网络是全局属性还是本地属性 封装库1、原理图中器件的封装与手册一致。 2、原理图器件是否是标准库的symbol。 绘制要求原理图中器件的封装与手册一致。 指示灯设计默认由电源点亮的指示灯和由MCU点灭的指示灯,便于故障时直观判断电源问题还是MCU问题 网口连接 器 确认网口连接器的开口方向、是否带指示灯以及是否带PoE 网口变压 器 确认变压器选型是否满足需求,比如带PoE 按键确认按键型号是直按键还是侧按键 电阻上下 拉 同一网络避免重复上拉或者下拉 OD门芯片的OD门或者OC门的输出管脚需要上拉
匹配高速信号的始端和末端需要预留串阻 三极管三极管电路需要考虑通流能力 可测试性在单板的关键电路和芯片附近增加地孔,便于测试 连接器防 呆 连接器选型时需要选择有防呆设计的型号 仿真低速时钟信号,一驱动总线接口下挂器件的驱动能力、匹配方式、接口时序必须经过仿真确认,例如MDC/MDIO、IIC、PCI、Local b us 仿真电路中使用电感、电容使用合适Q值,可以通过仿真。时序确认上电时序是否满足芯片手册和推荐电路要求。 时序确认下电时序是否满足芯片手册和推荐电路要求。 时序确认复位时序是否满足芯片手册和推荐电路要求。 复位开关单板按键开关设计,要防止长按按键,单板挂死问题,建议按键开关设计只产生一段短脉宽低电平。 复位设计复位信号设计 (1)依据芯片要求进行上下拉 (2)确认芯片复位的默认状态 (3)Peset信号并联几十PF的电容滤波,优化信号质量。(4)复位信号保证型号完整性。 复位所有接口和光模块默认处于复位状态。 电平匹配不同电平标准互连,关注电压、输入输出门限、匹配方式。 功耗详细审查各个芯片的功耗设计,计算出单板各个电压的最大功耗,选择有一定余量的电源。 缓启热插拔电路要进行缓启动设计 磁珠小电压大电流(安培级)值电源输出端口的磁珠,需要考虑磁珠压降 连接器板间电源连接器通流能力及压降留有预量 标识扣板与母板插座网络标识是否一致,前后插卡连机器管脚信号要一一对应。
汽车冲压件模具验收CHECKLIST
汽车冲压件模具验收CHECKLIST (转载1/2) 2010-10-13 10:18:26| 分类:默认分类| 标签:|字号大中小订阅 功能检查项目检查方式及要求 常规 产品如图示(对称) 工艺图的内容全部完成工艺提供 基准点尺寸- 平面图 基准点尺寸- 剖面图 模架尺寸符合压机图对于选定的主辅生产线,模架的安装性与压机匹配活动部分的行程工作行程符合图纸要求 弹簧符合行程测算弹簧行程,选择合适弹簧;对于使用氮气缸,要求能够提供使用依据及使用的压力值 氮气弹簧的固定检查氮气弹簧与安装面和顶料面是否垂直,误差小于1.5度,用直角尺和塞尺个弹簧进行 检查,根据情况选用合适的安装方式,对于不合适的安装方式,坚决纠正,每个气弹簧必须 标注实际压力值,请进行抽查。 部品的加工性尽量选用标准件,以便于损坏后加工更换 磨损件的装配性易损件安装拆换必须方便 易被损坏的部件用钢板保护模具周边露在外表部分易损件用保护罩挡,并刷红漆标示 无尖锐部件模具周边及其部件(特殊要求除外)必须全部倒角和R角 铸件之外的材料各部件必须有明显的材料牌号,零件编号及热处理硬度标示 材料表检查图纸的目录,每个部件均要有材料标示 氮气弹簧和普通弹簧不混用禁止将氮气弹簧与其它形式的弹簧混用在同一顶料和压料部件上镶块的重量应在部件上标明重量、硬度、编号 确认标准件根据工艺设计要求,按图纸标准检验 预压弹簧的润滑中间螺栓要有黄油润滑 非标件的详图所有非标件要求提供图纸,并检查非标件与图纸的符合性 三销孔- 上下型至少有三个加工基准孔(即三销孔),检查三个加工基准孔与设计是否一致,注孔的精度情况,孔径内不能有油漆,检查三销孔的孔径是否标准,并标注坐标尺 三销孔- 凸模 三销孔- 压料板 三销孔- 活动铸件 整形区域无孔在整形工作面上无铸造沙眼及其它缺陷孔 到底标记和产品标记的位置及有效G2线上的模具至少两个到底标记,位置要合理,不应在零件的表面和圆弧变形有效(标记0.2)。高出凹模型面约0.2mm,零件上的压印痕迹必须是圆周的以上。 计算力量 工作部件的行程关系图 填充型面的斜锲先于压料板运动检查斜锲各个压料板是否正常合理,测导向及压料板间隙在0.02~0.05mm之活回位,如果有3个以上滑动导向板,注意检查各导向板上下是否能够正常接触塞尺和红丹进行检察) 如果部件不平衡,设置反侧为了保持平衡,增设反侧装置,检查反侧装置的间隙(正常工作情况反侧之间无隙) 在压机下的安装 模具闭合高度按设计要求模具闭合高度为1240误差0~1mm,平面度要求每米0.1mm误闭合模具进入压机的高度模具高度不能超过侧门高度,根据压机参数设定 闭合模具进入压机尺寸 下模尺寸
英特尔平台 硬件设计入门指南
1. 需求分析及前期准备 1). 详细研究并理解实际需求; 如:性能指标、产品功能要求、机构认证需求、项目成本目标等,明确设计任务; 2). 学习Intel平台资料,针对设计任务和要求,设计可靠合理、经济可行的设计文档,进行评估; 需要特别注意Intel 平台各项功能参数与实际需求相符合;具体参考&中文产品说明网站,进行芯片选型; 3). 需要时联系Intel 工程师了解产品路线图,讨论芯片选型,开发调试工具(ITP etc.),项目开发计划及时间表,或申请Intel CRB 调试评估方案,缩短产品开发周期,避免走弯路; 4). 参考Intel 平台各项功能参数&EDS, 内说明特性指标章节; 5). 需要产品定义初期研究讨论软件实现的可行性,如与BIOS厂家讨论BIOS开发与设计,EC的配合程度,OS 与driver配合程度; 2. 可行性分析及设计文档 1). 务必画出系统架构/线路框图/电源分配/时钟/上电时序与复位,中断,调试等设计单元框图,并且深刻理解; 2). 依据Intel平台原理框图,进行器件选择与单元设计,以及EC方案的制定; 3). 注意CPU &chipset的工作电压、工作频率、时钟、时序、功耗等,满足设计需求; 4). 参考资料:DG,EDS,CRB 3. 硬件设计之原理图 1). 绘制原理图时排版清晰合理、排列均匀,可参考Intel原理图库文件Lib; 2). 建议参考Intel CRB 线路设计,注意电源分配,时钟安排,高速信号的连接等; 3). 认真与Intel 的schematic design check list排查容易出错的地方,尤其是DDR、PCIE等高速信号; 4). 需要及时与BIOS工程师讨论准备好BIOS,为第一版打样与试产的开机作好准备; 5). 原理图设计小组最终讨论并修改,审查项目:功能/性能/冗余设计/DFx(可生产性、可调试性、可测试性); 6). 参考资料:DG,EDS,CRB, Schematic Design Check List 4. 硬件设计之PCB布局 1). 与机构/外观/EMI/Power/RF/thermal team 讨论主要芯片的位置摆放,以满足产品设计方案需求; 2).计算各组总线走线宽度、讨论电源元器件位置及走线、敏感元器件位置摆放,产生正式设计文档; 3). I/O 端口位置,温度,时钟等元器件位置讨论,产生正式设计文档; 4). 参考资料:DG,CRB file. 5. 硬件设计之PCB布线 1). 绘制PCB layout时选择叠层合理、元器件排列均匀,高速信号布线顺畅,严格遵守Intel参考设计文档; 2). 要注意干扰源及敏感信号的屏蔽、各种不同功能模块的供电要做到相对隔离; 3). 参考layout(customer reference board file)及layout库文件;注意电源分配、时钟安排、DDR等高速信号的连接; 4). 认真与layout check list一项一项的排查,尤其是高速信号、电源、EMI对策等部分; 5). 在底片发给PCB板厂制板前,进行全面layout check, gerber out check 会议(审查项目:如SI/PI/DFx),讨论并修改后产生正式设计文档; 6). 参考:DG,CRB file, layout checklist,TLC:Trace Length Calculator.,
相关文档
- (完整版)汽车仪表板checklist
- 电路设计checklist
- PPAP检查清单Checklist(中英文)
- 结构设计检查表checklist
- Tool Buy-off Checklist
- 代码自查checklist
- checklist
- 汽车内外饰工艺数据checklist
- 代码大全核心checklist
- 汽车组合仪表校核规范checklist
- AUDIT Checklist(查检表) 波焊
- 汽车副仪表板(Console)设计限制面介绍
- 汽车通用提车检查表checklist
- ASES-Checklist(中日文对照版)
- 产品结构设计等方面的checklist0204192335
- ASES_Checklist(中日文对照版)
- BIQS-Audit-Checklist(中英文)
- 工艺总体方案Checklist
- 钣金结构设计CHECKLIST
- IPQC Checklist