如何利用BioEdit和Clustalx进行序列比对

1、将要进行序列比对的信息保存为.txt文本文档类型(如11.txt和21.txt)
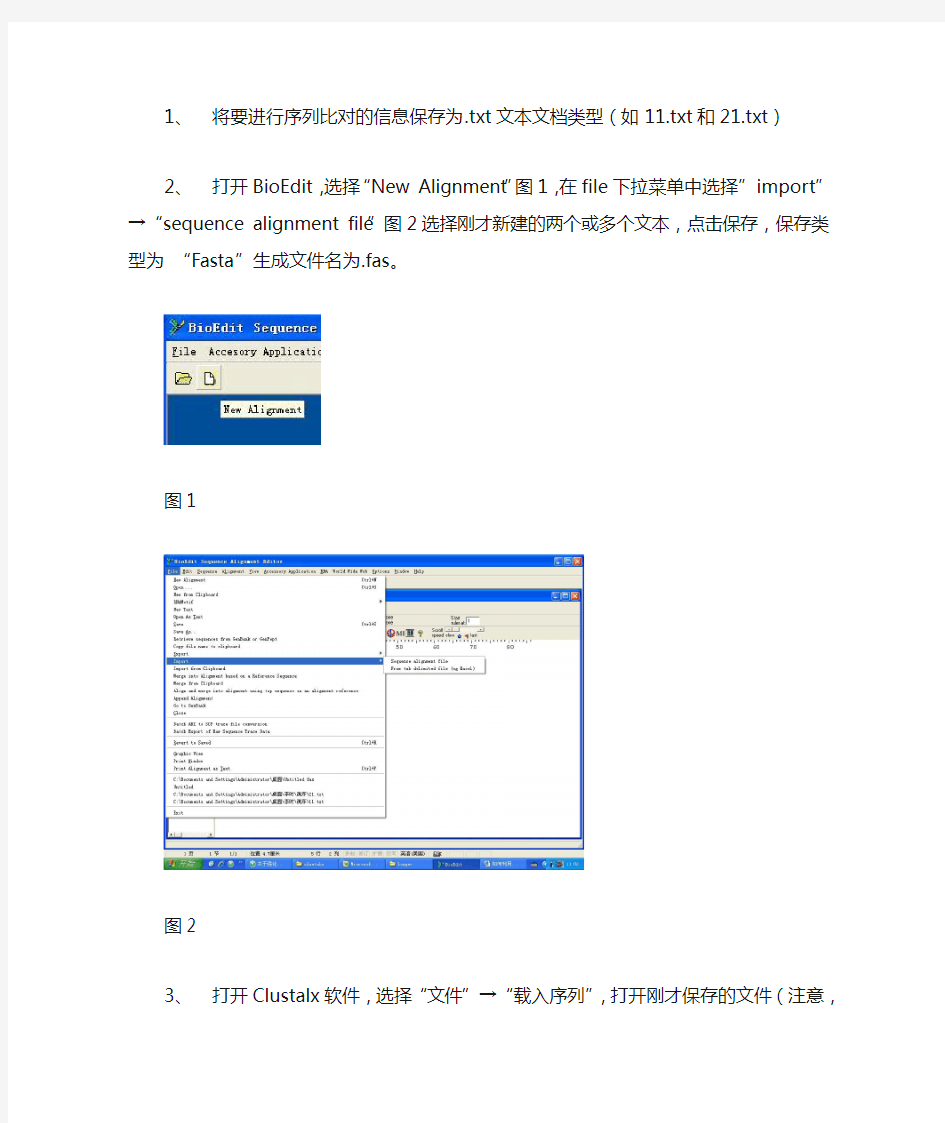
2、打开BioEdit,选择“New Alignment”图1,在file下拉菜单中选择”import”→“sequence
alignment file”图2选择刚才新建的两个或多个文本,点击保存,保存类型为“Fasta”
生成文件名为.fas。
图1
图2
3、打开Clustalx软件,选择“文件”→“载入序列”,打开刚才保存的文件(注意,该文
件应该与ClustalX在同一文件夹中),选择“序列比对”→“完全序列比对”,这时会生成两个文件,.aln和.dnd文件,其中.aln为序列比对文件,而.dnd为树文件。
4、打开BioEdit软件,选择选择“New Alignment”,在file下拉菜单中选择”import”→
“sequence alignment file”找到刚才生成的.aln文件打开就可以进行比对分析了。5、
多重序列比对及系统发生树的构建
多重序列比对及系统发生树的构建 作者:佚名来源:生物秀时间:2007-12-31 【实验目的】 1、熟悉构建分子系统发生树的基本过程,获得使用不同建树方法、建树材料和建树参数对建树结果影响的正确认识; 2、掌握使用Clustalx进行序列多重比对的操作方法; 3、掌握使用Phylip软件构建系统发生树的操作方法。 【实验原理】 在现代分子进化研究中,根据现有生物基因或物种多样性来重建生物的进化史是一个非常重要的问题。一个可靠的系统发生的推断,将揭示出有关生物进化过程的顺序,有助于我们了解生物进化的历史和进化机制。 对于一个完整的进化树分析需要以下几个步骤:⑴要对所分析的多序列目标进行比对(alignment)。⑵要构建一个进化树(phyligenetic tree)。构建进化树的算法主要分为两类:独立元素法(discrete character methods)和距离依靠法(distance methods)。所谓独立元素法是指进化树的拓扑形状是由序列上的每个碱基/氨基酸的状态决定的(例如:一个序列上可能包含很多的酶切位点,而每个酶切位点的存在与否是由几个碱基的状态决定的,也就是说一个序列碱基的状态决定着它的酶切位点状态,当多个序列进行进化树分析时,进化树的拓扑形状也就由这些碱基的状态决定了)。而距离依靠法是指进化树的拓扑形状由两两序列的进化距离决定的。进化树枝条的长度代表着进化距离。独立元素法包括最大简约性法(Maximum Parsimony methods)和最大可能性法(Maximum Likelihood methods);距离依靠法包括除权配对法(UPGMAM)和邻位相连法(Neighbor-joining)。⑶对进化树进行评估,主要采用Bootstraping法。进化树的构建是一个统计学问题,我们所构建出来的进化树只是对真实的进化关系的评估或者模拟。如果我们采用了一个适当的方法,那么所构建的进化树就会接近真实的“进化树”。模拟的进化树需要一种数学方法来对其进行评估。不同的算法有不同的适用目标。一般来说,最大简约性法适用于符合以下条件的多序列:i 所要比较的序列的碱基差别小,ii 对于序列上的每一个碱基有近似相等的变异率,iii 没有过多的颠换/转换的倾向,iv 所检验的序列的碱基数目较多(大于几千个碱基);
多序列比对
在寻找基因和致力于发现新蛋白的努力中,人们习惯于把新的序列同已知功能的蛋白序列作比对。由于这些比对通常都希望能够推测新蛋白的功能,不管它们是双重比对还是多序列比对,都可以回答大量的其它的生物学问题。举例来说,面对一堆搜集的比对序列,人们会研究隐含于蛋白之中的系统发生的关系,以便于更好地理解蛋白的进化。人们并不只是着眼于某一个蛋白,而是研究一个家族中的相关蛋白,看看进化压力和生物秩序如何结合起来创造出新的具有虽然不同但是功能相关的蛋白。研究完多序列比对中的高度保守区域,我们可以对蛋白质的整个结构进行预测,并且猜测这些保守区域对于维持三维结构的重要性。 显然,分析一群相关蛋白质时,很有必要了解比对的正确构成。发展用于多序列比对的程序是一个很有活力的研究领域,绝大多数方法都是基于渐进比对(progressive alignment)的概念。渐进比对的思想依赖于使用者用作比对的蛋白质序列之间确实存在的生物学上的或者更准确地说是系统发生学上的相互关联。不同算法从不同方面解决这一问题,但是当比对的序列大大地超过两个时(双重比对),对于计算的挑战就会很令人生畏。在实际操作中,算法会在计算速度和获得最佳比对之间寻求平衡,常常会接受足够相近的比对。不管最终使用的是什么方法,使用者都必须审视结果的比对,因为再次基础上作一些手工修改是十分必要的,尤其是对保守的区域。 由于本书偏重于方法而不是原理,这里只讨论一小部分现成的程序。我们从两个多序列比对的方法开始,接下去是一系列的利用蛋白质家族中已知的模体或是式样的方法,最后讨论两个具有赠送的方法,因为绝大多数公开的算法不能达到出版物的数量。在本章结尾部分将会列出更详细的多序列比对的算法。 渐进比对方法 CLUSTAL W CLUSTAL W算法是一个最广泛使用的多序列比对程序,在任何主要的计算机平台上都可以免费使用。这个程序基于渐进比对的思想,得到一系列序列的输入,对于每两个序列进行双重比对并且计算结果。基于这些比较,计算得到一个距离矩阵,反映了每对序列 Bioinformatics: A Practical Guide to the Analysis of genes and Proteins Edited by A.D. Baxevanis and B.E.E. Ouellette ISBN 0-471-191965. pages 172-188. Copyright ? 1998 Wiley – Liss. Inc.