PCR设计引物时酶切位点的保护碱基表Cleavage Close to the End of DNA Fragments2
shRNA设计原则
shRNA实验成功不得不看的shrna 设计原则大总结 RNAi由于可以特异地使基因沉默或表达量降低而成为生物实验的强有力工具。其中shrna设计在慢病毒的构建上应用颇广,本文对shRNA设计原则和操作技巧进行介绍。1.克隆到shRNA表达载体中的shRNA包括两个短反向重复序列,中间由一茎环( [shRNA shrna 设计原则寡核苷酸酶切位点聚合酶] RNAi由于可以特异地使基因沉默或表达量降低而成为生物实验的强有力工具。其中shrna设计在慢病毒的构建上应用颇广,本文对shRNA设计原则和操作技巧进行介绍。 1.克隆到shRNA表达载体中的shRNA包括两个短反向重复序列,中间由一茎环(loop)序列分隔的,组成发夹结构,由polⅢ启动子控制。随后在连上5-6个T作为RNA聚合酶Ⅲ的转录终止子。 2.两个互补的寡核苷酸两端须带有限制性酶切位点。 3.StrataGENE发现29个寡核苷酸较之原先推荐的23个寡核苷酸可以更有效的抑制目的基因。 4.在启动子下游的酶切位点下方紧连一个C,使插入片段和启动子有一定空间间隔以确保转录的发生。 5.ShRNA目的序列的第一个碱基必须是G以确保RNA聚合酶转录。如果选择的目的序列不以G开头,必须在紧连正义链的上游加一个G。 6.ShRNA插入片段中的茎环应当靠近寡核苷酸的中央。不同大小和核苷酸序列的茎环都被成功的运用过。其中包含一个独特的限制性酶切位点的茎环利于检测带有shRNA插入片段的克隆。在比较了众多不同长度和序列的茎环,5'TCAAGAG3'序列最为有效(AMBION use)。 7.5-6个T必须放置在shRNA插入片段尾部以确保RNA聚合酶III终止转录(stop)。 8.在正义链和反义链序列上不能出现连续3个或以上的T。这可能导致shRNA
设计引物如何设计酶切位点
经常有战友对一些常见问题在丁香园反复问答了很多遍,所以希望园子中一些战友,特别是低分与0分战友,能将好的帖子归纳总结了一下,并结合自己的经验整理,一方面这是个学习提高的过程,另一方面也能帮助大家解决这方面的问题。同时如有不当或不完善的地方,希望各位战友不断补充,争取有朝一日我们能把园子里战友的经验系统整理,给大家以帮助。 我先把设计引物如何设计酶切位点这方面的帖子整理一下,因为昨天一下子看到三个相似问题。原帖如下:我想向你求教一个问题,假如说我想把胰岛素基因和腺病毒载体连接起来,如何确定设计目的基因PCR时的引物呢?和相应的限制性核酸内切酶呢?谢谢老师能给予讲解,谢谢 [整理]:最初的时候,由于害怕设计酶切位点最后切不开,所以经常采用最通用的方法,用T载体克隆解决问题,但后来发现她也有问题,就是浓度提不上去,你需要体大量的载体来酶切,所以感到还是直接扩增好一点。但这就需要你仔细设计引物。连入质粒中的重要目的就是进行酶切和连接,当然首先就是在想要合成或者是进行PCR扩增出靶基因的时候在核酸的两端接入酶切位点,酶切位点是与你的质粒的特点相关的,可以在质粒的图谱说明书上找取相应的位点,进行设计。 (一)设计引物前应做的准备工作: 准备载体图谱,大致准备把片断插在那个部分 对片断进行酶切分析,确定一下那些酶切位点不能用 准备一本所买公司的酶的商品目录,便于查酶的各种数据及两种酶是否可以配用(二)设计引物所要考虑的问题 两个位点应是载体上的,,所连接片断上没有这两个位点,且距离不能太近,往往导致两个酶都切不好。因此,紧挨在一起,只能切一个,除非恰好是与上面两个酶在一起的酶切位点。我看promega的说明书上说,最好隔四个。还有一种情况是:不能有碱基的交叉,比如AGATCTTAAG,这样的位点比较难切。 两个酶切点最好不要是同尾酶(切下来的残基不要互补),否则效果相当于单酶切。 最好使用酶切效率高的。 最好使用双酶切有共同buffer的酶。
PCR设计引物时酶切位点的保护
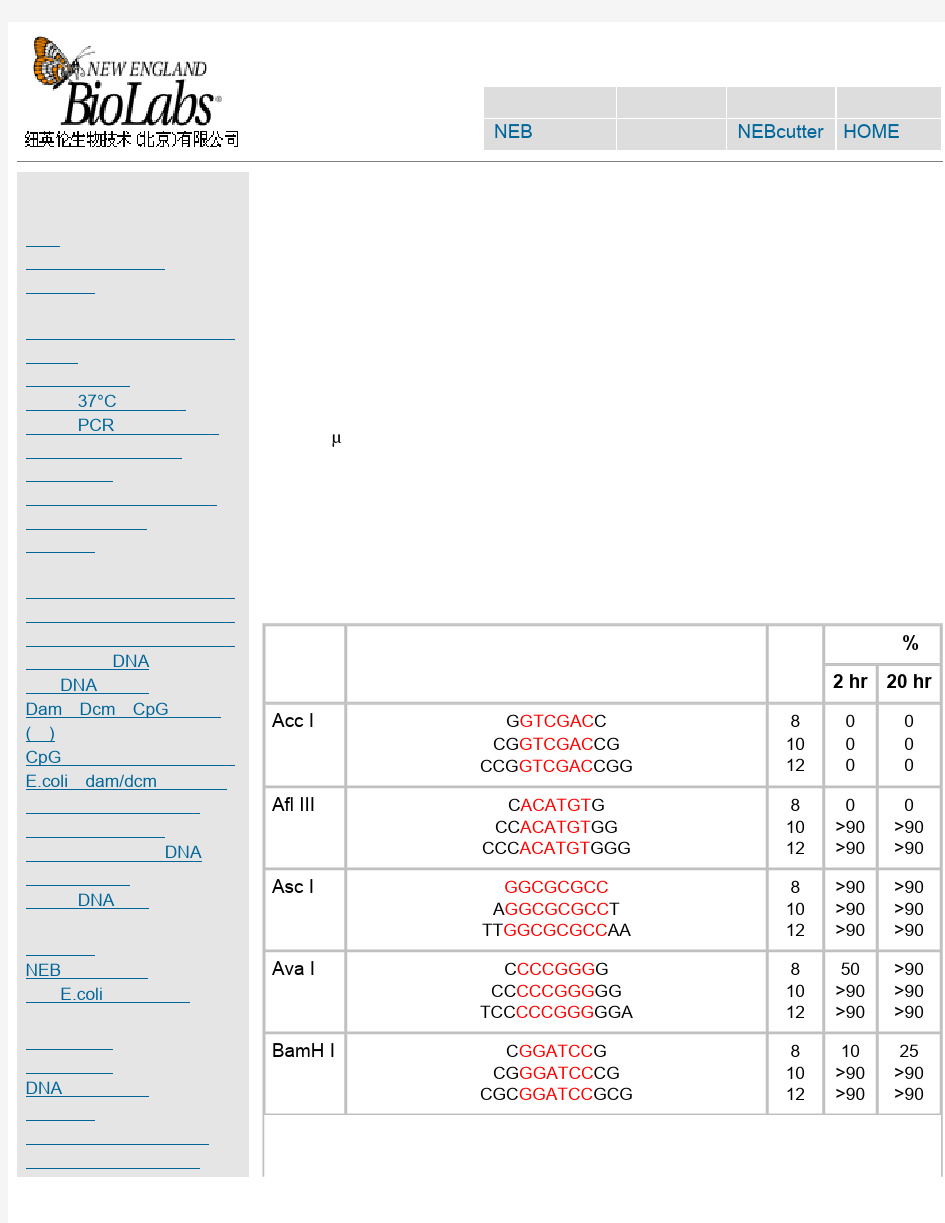
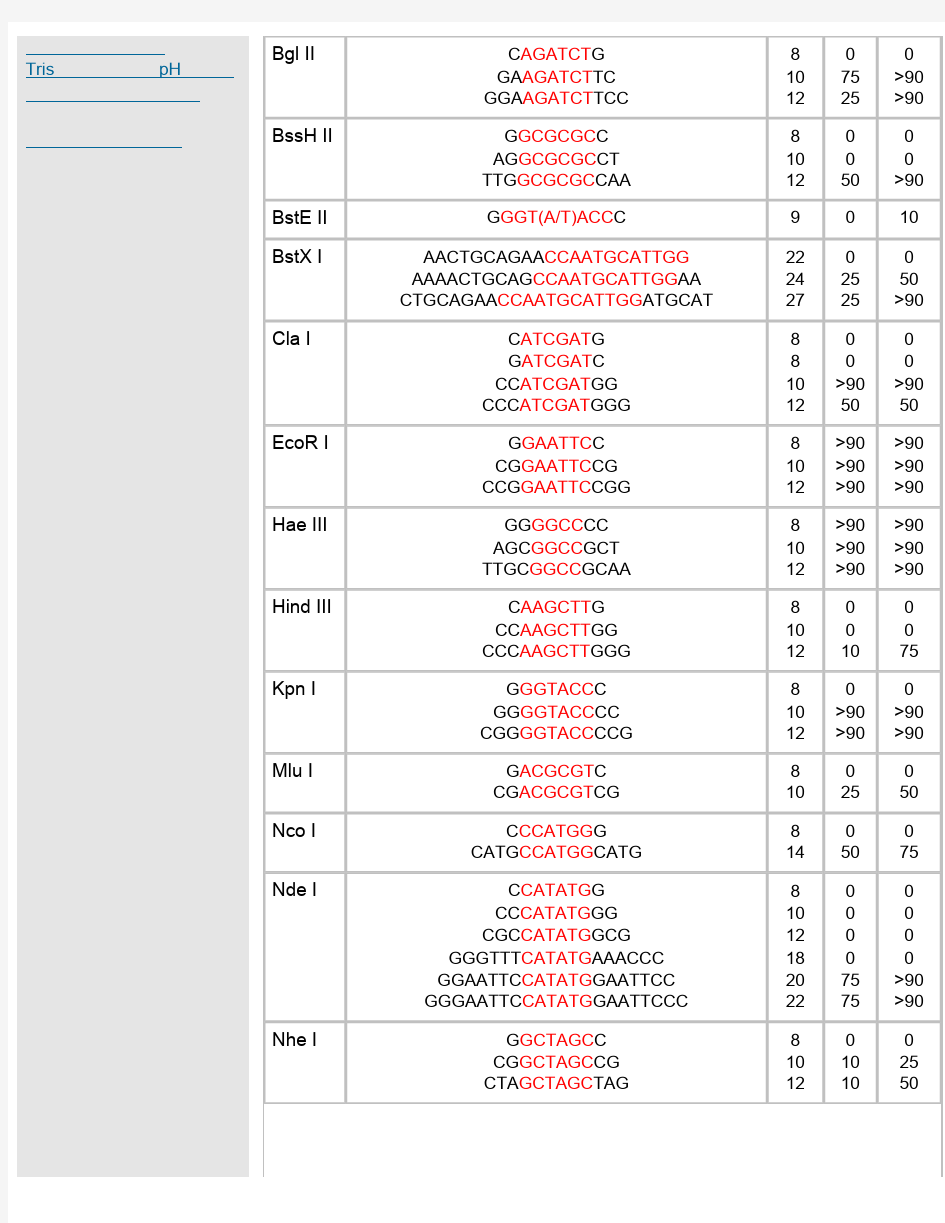
注释: 1.如果要加在序列的5‘端,就在酶切位点识别碱基序列(红色)的5’端加上相应的碱基(黑色),相同如果要在3‘端加保护碱基,就在酶切位点识别碱基序列(红色)的3’端加上相应的碱基(黑色)。 2.切割率:正确识别并酶切的效率 3。加保护碱基时最好选用切割率高时加的相应碱基。 为什么要添加保护碱基? 在分子克隆实验中,有时我们会在待扩增的目的基因片段两端加上特定的酶切位点,用于后续的酶切和连接反应。由于直接暴露在末端的酶切位点不容易直接被限制性核酸内切酶切开,因此在设计PCR引物时,人为的在酶切位点序列的5‘端外侧添加额外的碱基序列,即保护碱基,用来提高将来酶切时的活性。其次,在分子克隆实验中选择载体的酶切位点时,相临的两个酶切位点往往不能同时使用,因为一个位点切割后留下的碱基过少以至于影响旁边的酶切位点切割。 该如何添加保护碱基? 添加保护碱基时,最关心的应该是保护碱基的数目,而不是种类。什么样的酶切位点,添加几个保护碱基,是有数据可以参考的。 添加什么保护碱基,如果严格点,是根据两条引物的Tm值和各引物的碱基分布及GC含量。如果某条引物Tm值偏小,GC%较低,添加时多加G或C,反之亦反。为了解不同内切酶对识别位点以外最少保护碱基数目的要求,NEB采用了一系列含识别序列的短双链寡核苷酸作为酶切底物进行实验。实验结果对于确定双酶切顺序将会有帮助(比如在多接头上切割位点很接近时),或者当切割位点靠
近DNA末端时也很有用。在本表中没有列出的酶,则通常需在识别位点两端至少加上6个保护碱基,以确保酶切反应的进行。 实验方法:用γ-[32P]ATP在T4多聚核苷酸激酶的作用下标记0.1A260单位的寡核苷酸。取1?g已标记了的寡核苷酸与20单位的内切酶,在20°C条件下分别反应2小时和20小时。反应缓冲液含70mM Tris-HCl (pH 7.6), 10 mM MgCl2 , 5 mMDTT及适量的NaCl或KCl(视酶的具体要求而定)。20%的PAGE(7M尿素)凝胶电泳分析,经放射自显影确定酶切百分率。 本实验采用自连接的寡核苷酸作为对照。若底物有较长的回文结构,切割效率则可能因为出现发夹结构而降低。
引物设计原则及酶切位点选择和设计
引物设计原则及酶切位点选择和设计:最初的时候,由于害怕设计酶切位点最后且不开,所以经常采用最通用的方法,用[整理]载体克隆解决问题,但后来发现她也有问题,就是浓度提不上去,你需要体大量的载体来T连入质粒中的重要目的酶切,所以感到还是直接扩增好一点。但这就需要你仔细设计引物。扩增出靶基因的时候在核就是进行酶切和连接,当然首先就是在想要合成或者是进行PCR可以在质粒的图谱说明书上酸的两端接入酶切位点,酶切位点是与你的质粒的特点相关的,找取相应的位点,进行设计。(一)设计引物前应做的准备工作:准备载体图谱,大致准备把片断插在那个部分对片断进行酶切分析,确定一下那些酶切位点不能用准备一本所买公司的酶的商品目录,便于查酶的各种数据及两种酶是否可以配用(二)设计引物所要考虑的问题往往导致两个,所连接片断上没有这两个位点,且距离不能太近,两个位点应是载体上的,除非恰好是与上面两个酶在一起的酶切位点。只能切一个,酶都切不好。因此,紧挨在一起,还有一种情况是:不能有碱基的交叉,比如promega的说明书上说,最好隔四个。我看AGATCTTAAG,这样的位点比较难切。 两个酶切点最好不要是同尾酶(切下来的残基不要互补),否则效果相当于单酶切。最好使用酶切效率高的。的酶。最好使用双酶切有共同buffer最好使用自己实验室有的酶,这样可,ecor1等),最好使用较常用的酶(如hind3,bamh1以省钱。的问题,很多的战友都有疑惑。其实园子里有很多的解释了。的计算,关于TmTm大家可以理解,双链溶解所需的温度。即是DNA叫溶解温度(melting temperature, Tm),Tm因此,的溶解是没有作用的。而不互补的区域对DNA 这个温度是由互补的DNA区域决定的,(除时,只计算互补的区域Tm才有贡献。计算Tm只有和模板互补的区域对对于引物的Tm,过低,是因为他们误把保护碱。不少战友设计的引物都Tm 非你的酶切位点也与模板互补)反应的诸多困难。所以,设计引里了,最后的结果是导致了PCR 基和酶切位点都计算到Tm以度以上(我喜欢控制在58Tm控制在55物的时候,先不管5'端的修饰序列,把互补区的,再加上酶切位点和保Tm)的具体情况,对于困难的PCR,需要适当提高上,具体根据PCR温度高的引物就Tm护碱基,这样的引物通常都是可用的,即使有小的问题,也可以挽回。的公式。2+43'非特异结合等问题。简单的计算公式可以用比较容易克服3‘发卡、二聚体及,不包括酶切位点。引物公司给你发的单子是包括酶切位点90 若你计算的Tm值达到了快个碱基,去3329、的。自己可以再估计一下。如你设计了带酶切位点的引物,总长分别为多度,实际用的只7021个碱基。引物公司给的单子是掉酶切位点和保护碱基,分别为17、55度扩的结果也差不多。有50度,用其它关于Tm值的计算,有用PP5.0进行评价的,需要考虑的参数包括:base number、GC%、Tm、hairpin、dimer、false priming、cross dimer。退一般退火温度为Tm-5度,退火温度的计算可以不把加入的酶切位点及保护碱基考虑进去,如上所言,PCR 几个循环后,引物外侧的序列已经参入了扩增片断中,所以你可以在预变性后多加几步,温度比你Tm值低些(这样可能会增加非特异性),Tm值是你包括酶切位点及保护碱基的Primer计算出来的。1.一般在5'端加保护碱基,如果你扩增后把目的条带做胶回收转入T-VECTOR或者其它的载体的话,酶切时可以不需加保护碱基2.有人的经验加入酶切位点的引物可以和未加入时使用相同的退火结果也还是令人满意。,温度. 或是其他设计引物的软件进行计算一下,看看引物之间的关于引物二聚体,最好用primer时可以提高一下退△G(自由能)的绝对值,如果小于10,一般是问题的。如果稍大,PCR没火温度,一般是没有问题的。如果3'端形成二聚体,并且自由能绝对值较大,如果PCR 有条带,建议重新设计引物。。此外,所加的三个核苷酸的保护序列经过尽心设计有时也可以降低二聚体的△G一个特异性引物一在设计酶切位点时,最好能尽可能多的利用引物本身的碱基。这是因为,左右了。而我们在般都是20bp左右,再加上酶切位点序列和保护性碱基,大致就是28bp如果我
引物设计心得、Primer5.0 使用
引物设计、选择核酸内切酶、酶切位点之Primer5.0使用 下面以目的基因CD63 为例,介绍一下真核表达载体PcDNA3.1(+)-CD63的构建过程 1 cd63基因序列的获取 打开NCBI-选择 输入cd63 回车 找到第16条编码为 1 的 打开后可见该积基因的详细信息,找到CDS(编码序列)
可见其编码序列为第146到862位碱基,继续往下拉,可见基因序列,选中编码序列(146-862)复制,打开Primer 5.0 (起始密码子)ATG gc ggtggaagga ggaatgaagt gtgtcaagtt 181 tttgctctac gttctcctgc tggccttctg cgcctgtgca gtgggattga tcgccattgg 241 tgtagcggtt caggttgtct tgaagcaggc cattacccat gagactactg ctggctcgct 301 gttgcctgtg gtcatcattg cagtgggtgc cttcctcttc ctggtggcct ttgtgggctg 361 ctgtggggcc tgcaaggaga actactgtct catgattaca tttgccatct tcctgtctct 421 tatcatgctt gtggaggtgg ctgtggccat tgctggctat gtgtttagag accaggtgaa 481 gtcagagttt aataaaagct tccagcagca gatgcagaat taccttaaag acaacaaaac 541 agccactatt ttggacaaat tgcagaaaga aaataactgc tgtggagctt ctaactacac 601 agactgggaa aacatccccg gcatggccaa ggacagagtc cccgattctt gctgcatcaa 661 cataactgtg ggctgtggga atgatttcaa ggaatccact atccataccc agggctgcgt 721 ggagactata gcaatatggc taaggaagaa catactgctg gtggctgcag cggccctggg 781 cattgctttt gtggaggtct tgggaattat cttctcctgc tgtctggtga agagtattcg 841 aagtggctat gaagtaatg t ag 其中 tag((UAG)终止密码 子,设计引物的时候需要将其删除,因为,真核表达载体上多含有标
各种酶切位点的保护碱基引物设计必看
各种酶切位点的保护碱基 酶不同,所需要的酶切位点的保护碱基的数量也不同。一般情况下,在酶切位点以外多出3个碱基即可满足几乎所有限制酶的酶切要求。在资料上查不到的,我们一般都随便加3个碱基做保护。 寡核苷酸近末端位点的酶切 (Cleavage Close to the End of DNA Fragments (oligonucleotides) 为了解不同内切酶对识别位点以外最少保护碱基数目的要求,NEB采用了一系列含识别序列的短双链寡核苷酸作为酶切底物进行实验。实验结果对于确定双酶切顺序将会有帮助(比如在多接头上切割位点很接近时),或者当切割位点靠近DNA末端时也很有用。在本表中没有列出的酶,则通常需在识别位点两端至少加上6个保护碱基,以确保酶切反应的进行。 实验方法:用γ-[32P]ATP在T4多聚核苷酸激酶的作用下标记0.1A260单位的寡核苷酸。取1 μg 已标记了的寡核苷酸与20单位的内切酶,在20°C条件下分别反应2小时和20小时。反应缓冲液含70 mM Tris-HCl (pH , 10 mM MgCl2 , 5 mM DTT及适量的NaCl或KCl(视酶的具体要求而定)。20%的PAGE(7 M尿素)凝胶电泳分析,经放射自显影确定酶切百分率。 本实验采用自连接的寡核苷酸作为对照。若底物有较长的回文结构,切割效率则可能因为出现发夹结构而降低。
2.双酶切的问题 参看目录,选择共同的buffer。其实,双酶切选哪种buffer是实验的结果,takara公司从1979年开始生产限制酶以来,做了大量的基础实验,也积累了很多经验,目录中所推荐的双酶切buffer 完全是依据具体实验结果得到的。 有共同buffer的,通常按照常规的酶切体系,在37℃进行同步酶切。但BamH I在37℃下有时表现出star活性,常用30℃单切。 两个酶切位点相邻或没有共同buffer的,通常单切,即先做一种酶切,乙醇沉淀,再做另一种酶切。 3.酶切底物DNA,切不开 1)底物DNA上没有相应的限制酶识别位点,或酶切位点被甲基化。 2)PCR引物的酶切位点前没有保护碱基或引物合成有误,致使没有正确的酶切位点存在。PCR产物酶切前尽量进行精制以更换buffer。由于PCR产物中带入的其它物质,会影响酶切,据报道,通常PCR产物的添加量占总反应体积25%以下没有问题。 3)酶切条件的确认,包括反应温度和反应体系等。同样的DNA,同样量,用不同的限制酶切情况可能不同,由于DNA的空间结构造成的。同样的DNA,不同的反应体系,酶切效果也可能不同,由于一些空间因素或不可测因素造成的。 4)公司出售的限制酶都是液体状态,都是根据最佳反应体系配制了浓度,不可以再用buffer稀释,因为酶浓度和活性之间不呈直线对应关系,酶浓度越稀,相对活性越低,并且越不稳定,有时便会出现底物DNA不能被切断的现象。 不同公司的酶和buffer不要交叉使用,否则可能会影响酶切效果。 5)酶的识别位点上的碱基被甲基化。可以选用不受甲基化影响的同裂酶,或将质粒DNA转入甲基化酶欠损的宿主菌中,重新制备DNA,也可以使用PCR的方法对DNA进行扩增,再做酶切。常用的有XbaI容易受甲基化影响,通常选用GM33做宿主菌转化。 6)底物不纯,含有限制酶阻害物质,影响酶切作用,需要重新纯化DNA。一般做乙醇沉淀纯化即可。如果质粒中含盐或酚等,都会影响酶切效果。
引物设计原则及酶切位点选择和设计
引物设计原则及酶切位点选择和设计 [整理]:最初的时候,由于害怕设计酶切位点最后且不开,所以经常采用最通用的方法,用T载体克隆解决问题,但后来发现她也有问题,就是浓度提不上去,你需要体大量的载体来酶切,所以感到还是直接扩增好一点。但这就需要你仔细设计引物。连入质粒中的重要目的就是进行酶切和连接,当然首先就是在想要合成或者是进行PCR扩增出靶基因的时候在核酸的两端接入酶切位点,酶切位点是与你的质粒的特点相关的,可以在质粒的图谱说明书上找取相应的位点,进行设计。 (一)设计引物前应做的准备工作: 准备载体图谱,大致准备把片断插在那个部分 对片断进行酶切分析,确定一下那些酶切位点不能用 准备一本所买公司的酶的商品目录,便于查酶的各种数据及两种酶是否可以配用 (二)设计引物所要考虑的问题 两个位点应是载体上的,,所连接片断上没有这两个位点,且距离不能太近,往往导致两个酶都切不好。因此,紧挨在一起,只能切一个,除非恰好是与上面两个酶在一起的酶切位点。我看promega的说明书上说,最好隔四个。还有一种情况是:不能有碱基的交叉,比如AGATCTTAAG,这样的位点比较难切。 两个酶切点最好不要是同尾酶(切下来的残基不要互补),否则效果相当于单酶切。 最好使用酶切效率高的。 最好使用双酶切有共同buffer的酶。 最好使用较常用的酶(如hind3,bamh1,ecor1等),最好使用自己实验室有的酶,这样可以省钱。 Tm的计算,关于Tm的问题,很多的战友都有疑惑。其实园子里有很多的解释了。 Tm叫溶解温度(melting temperature, Tm),即是DNA双链溶解所需的温度。大家可以理解,这个温度是由互补的DNA区域决定的,而不互补的区域对DNA的溶解是没有作用的。因此,对于引物的Tm,只有和模板互补的区域对Tm才有贡献。计算Tm时,只计算互补的区域(除非你的酶切位点也与模板互补)。不少战友设计的引物都Tm过低,是因为他们误把保护碱基和酶切位点都计算到Tm里了,最后的结果是导致了PCR反应的诸多困难。所以,设计引物的时候,先不管5'端的修饰序列,把互补区的Tm控制在55度以上(我喜欢控制在58以上,具体根据PCR的具体情况,对于困难的PCR,需要适当提高Tm),再加上酶切位点和保护碱基,这样的引物通常都是可用的,即使有小的问题,也可以挽回。Tm温度高的引物就比较容易克服3‘发卡、二聚体及3'非特异结合等问题。简单的计算公式可以用2+4的公式。若你计算的Tm值达到了快90 ,不包括酶切位点。引物公司给你发的单子是包括酶切位点的。自己可以再估计一下。如你设计了带酶切位点的引物,总长分别为29、33个碱基,去掉酶切位点和保护碱基,分别为17、21个碱基。引物公司给的单子是70多度,实际用的只有50度,用55度扩的结果也差不多。 其它关于Tm值的计算,有用PP5.0进行评价的,需要考虑的参数包括:base number、GC%、Tm、hairpin、dimer、false priming、cross dimer。退一般退火温度为Tm-5度,退火温度的计算可以不把加入的酶切位点及保护碱基考虑进去,如上所言,PCR几个循环后,引物外侧的序列已经参入了扩增片断中,所以你可以在预变性后多加几步,温度比你Tm值低些(这样可能会增加非特异性),Tm值是你包括酶切位点及保护碱基的Primer计算出来的。1.一般在5'端加保护碱基,如果你扩增后把目的条带做胶回收转入T-VECTOR或者其它的载体的话,酶切时可以不需加保护碱基2.有人的经验加入酶切位点的引物可以和未加入时使用相同的退火温度,结果也还是令人满意。
实验2-设计性实验-不同限制性内切酶对质粒切割-指导讲解
实验二不同限制性核酸内切酶对质粒DNA 的切割(设计性实验) 设计实验目的:培养学生独立完成实验设计、实验操作和撰写小论文的能力。 设计实验要求:选用1或2种内切酶,将所提供的质粒DNA切割为至少3-4个以上的片段。 设计实验过程:学生根据实验要求,运用学过的知识,先设计实验方案,提交老师批阅认可;再独立进行实验准备和实验;实验结束,撰写设计性实验报告,即小论文1篇。小论文要求:字数不少于3000,包括论文题目、作者、摘要、关键词、引言、材料与方法、结果与分析、结论或讨论、参考文献等。 设计实验涉及知识点:1)质粒的限制性内切酶图谱;2)质粒酶切鉴定;3)DNA片段的琼脂糖凝胶电泳。 设计实验要求学生独立撰写小论文。
Digestion of plasmid DNA with different restriction endonuclease 不同限制性核酸内切酶对质粒DNA的切割 一、原理 限制性内切酶能特异地结合于一段被称为限制性酶识别序列的DNA序列之内或其附近的特异位点上,并切割双链DNA。它可分为三类:Ⅰ类和Ⅲ类酶在同一蛋白质分子中兼有切割和修饰(甲基化)作用且依赖于A TP的存在。Ⅰ类酶结合于识别位点并随机的切割识别位点不远处的DNA,而Ⅲ类酶在识别位点上切割DNA分子, 然后从底物上解离。Ⅱ类由两种酶组成: 一种为限制性内切核酸酶(限制酶),它切割某一特异的核苷酸序列; 另一种为独立的甲基化酶, 它修饰同一识别序列。Ⅱ类中的限制性内切酶在分子克隆中得到了广泛应用,它们是重组DNA的基础。绝大多数Ⅱ类限制酶识别长度为4至6个核苷酸的回文对称特异核苷酸序列(如EcoRⅠ识别六个核苷酸序列:5'- G↓AATTC-3'),有少数酶识别更长的序列或简并序列。Ⅱ类酶切割位点在识别序列中,有的在对称轴处切割,产生平末端的DNA片段(如SmaⅠ: 5'-CCC↓GGG-3');有的切割位点在对称轴一侧,产生带有单链突出末端的DNA片段称粘性未端, 如EcoRⅠ切割识别序列后产生两个互补的粘性末端。 5'…G↓AATTC…3' →5'… G AATTC…3' 3'…CTTAA↑G …5' →3'… CTTAA G…5' 大部分限制性内切酶不受RNA或单链DNA的影响。当微量的污染物进入限制性内切酶贮存液中时, 会影响其进一步使用,因此在吸取限制性内切酶时, 每次都要用新的吸管头。如果采用两种限制性内切酶, 必须要注意分别提供各自的最适盐浓度。若两者可用同一缓冲液,则可同时水解。若需要不同的盐浓度, 则低盐浓度的限制性内切酶必须首先使用, 随后调节盐浓度,再用高盐浓度的限制性内切酶水解。也可在第一个酶切反应完成后,用等体积酚/氯仿抽提,加0.1倍体积3mol/L NaAc和2倍体积无水乙醇,混匀后置-70℃低温冰箱30分钟,离心、干燥并重新溶于缓冲液后进行第二个酶切反应。 DNA限制性内切酶酶切图谱又称DNA的物理图谱,它由一系列位置确定的多种限制性内切酶酶切位点组成,以直线或环状图式表示。本实验采用重组质粒pUC19-P35S:ARF8,其物理图谱如图1所示。
各种酶切位点的保护碱基引物设计必看
各种酶切位点的保护碱基引物设计必看 Document serial number【KK89K-LLS98YT-SS8CB-SSUT-SST108】
各种酶切位点的保护碱基酶不同,所需要的酶切位点的保护碱基的数量也不同。一般情况下,在酶切位点以外多出3个碱基即可满足几乎所有限制酶的酶切要求。在资料上查不到的,我们一般都随便加3个碱基做保护。 寡核苷酸近末端位点的酶切 (Cleavage Close to the End of DNA Fragments (oligonucleotides) 为了解不同内切酶对识别位点以外最少保护碱基数目的要求,NEB采用了一系列含识别序列的短双链寡核苷酸作为酶切底物进行实验。实验结果对于确定双酶切顺序将会有帮助(比如在多接头上切割位点很接近时),或者当切割位点靠近DNA末端时也很有用。在本表中没有列出的酶,则通常需在识别位点两端至少加上6个保护碱基,以确保酶切反应的进行。 实验方法:用γ-[32P]ATP在T4多聚核苷酸激酶的作用下标记0.1A260单位的寡核苷酸。取1 μg 已标记了的寡核苷酸与20单位的内切酶,在20°C条件下分别反应2小时和20小时。反应缓冲液含70 mM Tris-HCl (pH , 10 mM MgCl2 , 5 mM DTT及适量的NaCl或KCl(视酶的具体要求而定)。20%的PAGE(7 M尿素)凝胶电泳分析,经放射自显影确定酶切百分率。 本实验采用自连接的寡核苷酸作为对照。若底物有较长的回文结构,切割效率则可能因为出现发夹结构而降低。
2.双酶切的问题 参看目录,选择共同的buffer。其实,双酶切选哪种buffer是实验的结果,takara公司从1979年开始生产限制酶以来,做了大量的基础实验,也积累了很多经验,目录中所推荐的双酶切buffer完全是依据具体实验结果得到的。 有共同buffer的,通常按照常规的酶切体系,在37℃进行同步酶切。但BamH I在37℃下有时表现出star活性,常用30℃单切。 两个酶切位点相邻或没有共同 buffer的,通常单切,即先做一种酶切,乙醇沉淀,再做另一种酶切。 3.酶切底物DNA,切不开 1)底物DNA上没有相应的限制酶识别位点,或酶切位点被甲基化。 2)PCR引物的酶切位点前没有保护碱基或引物合成有误,致使没有正确的酶切位点存在。PCR产物酶切前尽量进行精制以更换buffer。由于PCR产物中带入的其它物质,会影响酶切,据报道,通常PCR产物的添加量占总反应体积25%以下没有问题。3)酶切条件的确认,包括反应温度和反应体系等。同样的DNA,同样量,用不同的限制酶切情况可能不同,由于DNA的空间结构造成的。同样的DNA,不同的反应体系,酶切效果也可能不同,由于一些空间因素或不可测因素造成的。
PCR酶切位点的设计
PCR酶切位点的设计 一、设计引物前应做的准备:准备载体图谱,大致准备把片段插在哪个部分,对片段进行酶切分析,确定哪些酶切位点不能用,准备一本公司酶的商品目录,便于查询各种酶的数据及两种酶是否可以配用。2.设计引物所要考虑的问题是两个位点应是载体上的,所以连接片段上没有这两个位点,且距离不能太近,往往导致两个酶都切不好。因此,紧挨在一起,只能切一个,除非恰好是与上面两个酶在一起的酶切位点,最好是隔4隔。两个酶切点最好不要是同尾酶(切下来的残基不要互补),否则效果相当于单酶切。最好使用酶切效率高的。最好使用双酶切有共同的buffer的酶。 二、Tm。设计引物的时候先不管5端的修饰序列,把互补区的T值控制在55度以上,在加上酶切位点和保护碱基。高的T值引物就比较容易克服3端发卡,二聚体及3’非特异结合等问题。 三、引物间的自由能的绝对值,如果小于10一般是问题不大的。如果稍大,PCR时可以提高一下退火温度,一般是没有问题的。如果3,端形成二聚体,并且自由能绝对值较大,如果PCR没有条带,建议重新设计引物。此外,所加的三个核苷酸的保护序列经过尽心设计有时候也可以降低二聚体的自由能。在设计酶切位点时最好能尽可能多的利用引物本身的碱基。这是因为一个特异性引物一般都是20bp左右,在加上酶切位点序列和保护碱基,大致就是28bp。 四、设计时限制性酶切位点应该是在5端的顶端。在设计引物时,常在5端添加酶切位点,以利于PCR产物连接到载体。设计引物时保证在最后5个核苷中含有3个A或T。先利用软件设计出合适的引物,引物的3端是引发延伸的起点,因此一定要与模板准确配对,应尽量避免在引物3端的第一位碱基是A.(容易错配)引物3端最佳碱基是G或C,行程的碱基比较稳定。 五、酶切位点都需要保护碱基,以利于内切酶的有效切割,酶切位点前加保护碱基1,两个酶切位点至少隔上3个碱基,在做载体构建的时候设计引物扩增片段进行定向连接,除了酶切位点,还要在两端加一个3个核算的保护序列,否则PCR产物很难被酶切,因此就会导致连接失败,因为内切酶需要一定的辅助性碱基才能顺利切割,在没有辅助碱基的情况下,有的酶是可以切割的,比如:Shali和SpeI,他们不需要辅助性碱基即可切割。但大部分酶是需要辅助性碱基的。
酶切位点的设计
酶切位点的设计 44 推荐 经常有战友对一些常见问题在丁香园反复问答了很多遍,所以希望园子中一些战友,特别是低分与0分战友,能将好的帖子归纳总结了一下,并结合自己的经验整理,一方面这是个学习提高的过程,另一方面也能帮助大家解决这方面的问题。同时如有不当或不完善的地方,希望各位战友不断补充,争取有朝一日我们能把园子里战友的经验系统整理,给大家以帮助。我先把设计引物如何设计酶切位点这方面的帖子整理一下,因为昨天一下子看到三个相似问题。原帖如下:我想向你求教一个问题,假如说我想把胰岛素基因和腺病毒载体连接起来,如何确定设计目的基因PCR时的引物呢?和相应的限制性核酸内切酶呢?谢谢老师能给予讲解,谢谢 [整理]:最初的时候,由于害怕设计酶切位点最后且不开,所以经常采用最通用的方法,用T载体克隆解决问题,但后来发现她也有问题,就是浓度提不上去,你需要体大量的载体来酶切,所以感到还是直接扩增好一点。但这就需要你仔细设计引物。连入质粒中的重要目的就是进行酶切和连接,当然首先就是在想要合成或者是进行PCR扩增出靶基因的时候在核酸的两端接入酶切位点,酶切位点是与你的质粒的特点相关的,可以在质粒的图谱说明书上找取相应的位点,进行设计。 (一)设计引物前应做的准备工作: 准备载体图谱,大致准备把片断插在那个部分 对片断进行酶切分析,确定一下那些酶切位点不能用 准备一本所买公司的酶的商品目录,便于查酶的各种数据及两种酶是否可以配用 (二)设计引物所要考虑的问题 两个位点应是载体上的,,所连接片断上没有这两个位点,且距离不能太近,往往导致两个酶都切不好。因此,紧挨在一起,只能切一个,除非恰好是与上面两个酶在一起的酶切位点。我看promega的说明书上说,最好隔四个。还有一种情况是:不能有碱基的交叉,比如AGATCTTAAG,这样的位点比较难切。 两个酶切点最好不要是同尾酶(切下来的残基不要互补),否则效果相当于单酶切。 最好使用酶切效率高的。 最好使用双酶切有共同buffer的酶。 最好使用较常用的酶(如hind3,bamh1,ecor1等),最好使用自己实验室有的酶,这样可以省钱。 Tm的计算,关于Tm的问题,很多的战友都有疑惑。其实园子里有很多的解释了。 Tm叫溶解温度(melting temperature, Tm),即是DNA双链溶解所需的温度。大家可以理解,这个温度是由互补的DNA区域决定的,而不互补的区域对DNA的溶解是没有作用的。因此,对于引物的Tm,只有和模板互补的区域对Tm才有贡献。计算Tm时,只计算互补的区域(除非你的酶切位点也与模板互补)。不少战友设计的引物都Tm过低,是因为他们误把保护碱基和酶切位点都计算到Tm里了,最后的结果是导致了PCR反应的诸多困难。所以,设计引物的时候,先不管5'端的修饰序列,把互补区的Tm控制在55度以上(我喜欢控制在58以上,具体根据PCR的具体情况,对于困难的PCR,需要适当提高Tm),再加上酶切位点和保护碱基,这样的引物通常都是可用的,即使有小的问题,也可以挽回。Tm温度高的引物就比较容易克服3‘发卡、二聚体及3'非特异结合等问题。简单的计算公式可以用2+4的公式。若你计算的Tm值达到了快90 ,不包括酶切位点。引物公司给你发的单子是包括酶切位点
酶切位点的设计
1.设计引物前应做的准备:准备载体图谱,大致准备把片段插在哪个部分,对片段进行酶 切分析,确定哪些酶切位点不能用,准备一本公司酶的商品目录,便于查询各种酶的数据及两种酶是否可以配用。2.设计引物所要考虑的问题是两个位点应是载体上的,所以连接片段上没有这两个位点,且距离不能太近,往往导致两个酶都切不好。因此,紧挨在一起,只能切一个,除非恰好是与上面两个酶在一起的酶切位点,最好是隔4隔。两个酶切点最好不要是同尾酶(切下来的残基不要互补),否则效果相当于单酶切。最好使用酶切效率高的。最好使用双酶切有共同的buffer的酶。 2.关于Tm。设计引物的时候先不管5端的修饰序列,把互补区的T值控制在55度以上, 在加上酶切位点和保护碱基。高的T值引物就比较容易克服3端发卡,二聚体及3,非特异结合等问题。 3.引物间的自由能的绝对值,如果小于10一般是问题不大的。如果稍大,PCR时可以提 高一下退火温度,一般是没有问题的。如果3,端形成二聚体,并且自由能绝对值较大,如果PCR没有条带,建议重新设计引物。此外,所加的三个核苷酸的保护序列经过尽心设计有时候也可以降低二聚体的自由能。在设计酶切位点时最好能尽可能多的利用引物本身的碱基。这是因为一个特异性引物一般都是20bp左右,在加上酶切位点序列和保护碱基,大致就是28bp。 4.设计时限制性酶切位点应该是在5端的顶端。在设计引物时,常在5端添加酶切位点, 以利于PCR产物连接到载体。设计引物时保证在最后5个核苷中含有3个A或T。先利用软件设计出合适的引物,引物的3端是引发延伸的起点,因此一定要与模板准确配对,应尽量避免在引物3端的第一位碱基是A.(容易错配)引物3端最佳碱基是G或C,行程的碱基比较稳定。 5.酶切位点都需要保护碱基,以利于内切酶的有效切割,酶切位点前加保护碱基1,两个 酶切位点至少隔上3个碱基,在做载体构建的时候设计引物扩增片段进行定向连接,除了酶切位点,还要在两端加一个3个核算的保护序列,否则PCR产物很难被酶切,因此就会导致连接失败,因为内切酶需要一定的辅助性碱基才能顺利切割,在没有辅助碱基的情况下,有的酶是可以切割的,比如:Shali和SpeI,他们不需要辅助性碱基即可切割。但大部分酶是需要辅助性碱基的。所以引物顺序应是5,-保护碱基+酶切位点+引物配对区---3,只要在5,端加保护碱基就可以了。其实保护碱基就是要给限制性内切酶一个结合位点,3端还有更长的引物序列存在。
引物设计方法
引物设计 正向引物:(1)首先从目的蛋白的基因序列的开头选取近十五个左右的基因,然后放入Tm Calculator (https://www.sodocs.net/doc/6416410229.html,/webtools/tmc/)中检测Tm值,是否在65℃左右。如果过低,则将取的基因数目增加,一直到Tm满足要求;如果过高,则减少所取的基因数目,满足Tm要求(bp 不能小于15bp,一般为15-30bp)。另外,5’端不能改变,3’端必须以C或G结尾。 (2)在质粒载体上找到最佳的酶切位点,不要切掉质粒载体中的有用部分或者标签(质粒载体有分N-端与C-端),选择好酶切位点。(3)使用ApE(软件,可下载),将目的蛋白的基因全部黏贴进去,检查其中所有的酶切位点(Enzyme——> Enzyme selector)。如果我们选择的酶切位点在目的基因中存在,则此酶切位点不能使用(会导致基因产物无法获得);若选择的酶切位点不存在与目的基因中,则可以使用。 (4)质粒载体中选取的酶切位点,将其基因序列放到NEBcutter (https://www.sodocs.net/doc/6416410229.html,/NEBcutter2/)中进行更加精确的酶切位点 显示(如:),再次确定。 (5)最后确定连接到引物上的酶切位点序列需要充分考虑ORF的
要求(即三个基因确定一个蛋白质,错位的话会导致目的产物错误),从ATG开始,不能发生错位。(例如:CTT GCG GCC GC G AAT TC A中)TCA 为一组,而酶切位点到TC结束,必须将A加上。然后从酶切位点开始,如上图即从AATTC中的T开始往左数16bp的基因(至少15bp,15-18bp)。然后将其连接到(1)中满足Tm的基因的5’端。(如:CTT GCG GCC GC G AAT TC A atggcg gatgaagcca c) 附:如果引物中酶切位点最后为ATG 而与其相连的目的蛋白的基因开始也是ATG 则可以删除一组,留一组即可。例:Nde1 Ufm 1 FOR CGCGCGGCAGC CATATG(atg)tcgaaggtttcctttaagatcac 反向引物:步骤与正向引物相似,不过(1)从目的蛋白的基因的3’-端开始往前取,取到满足Tm值的。其次,此时3’端不能变,而5’端必须以C或G结尾。(2)(3)(4)步骤都一样,(5)可以不用考虑ORF的问题,因为其在酶切位点就不会再往下pcr了。设计好后,使用ApE中的Reverse Complement,使其反转,完成引物设计。
酶切位点的设计
(一)设计引物前应做的准备工作: 准备载体图谱,大致准备把片断插在那个部分 对片断进行酶切分析,确定一下那些酶切位点不能用 准备一本所买公司的酶的商品目录,便于查酶的各种数据及两种酶是否可以配用 (二)设计引物所要考虑的问题 两个位点应是载体上的,,所连接片断上没有这两个位点,且距离不能太近,往往导致两个酶都切不好。因此,紧挨在一起,只能切一个,除非恰好是与上面两个酶在一起的酶切位点。我看promega的说明书上说,最好隔四个。还有一种情况是:不能有碱基的交叉,比如AGATCTTAAG,这样的位点比较难切。 两个酶切点最好不要是同尾酶(切下来的残基不要互补),否则效果相当于单酶切。 最好使用酶切效率高的。 最好使用双酶切有共同buffer的酶。 最好使用较常用的酶(如hind3,bamh1,ecor1等),最好使用自己实验室有的酶,这样可以省钱。 Tm的计算,关于Tm的问题,很多的战友都有疑惑。其实园子里有很多的解释了。 Tm叫溶解温度(melting temperature, Tm),即是DNA双链溶解所需的温度。大家可以理解,这个温度是由互补的DNA区域决定的,
而不互补的区域对DNA的溶解是没有作用的。因此,对于引物的Tm,只有和模板互补的区域对Tm才有贡献。计算Tm时,只计算互补的区域(除非你的酶切位点也与模板互补)。不少战友设计的引物都Tm 过低,是因为他们误把保护碱基和酶切位点都计算到Tm里了,最后的结果是导致了PCR反应的诸多困难。所以,设计引物的时候,先不管5'端的修饰序列,把互补区的Tm控制在55度以上(我喜欢控制在58以上,具体根据PCR的具体情况,对于困难的PCR,需要适当提高Tm),再加上酶切位点和保护碱基,这样的引物通常都是可用的,即使有小的问题,也可以挽回。Tm温度高的引物就比较容易克服3‘发卡、二聚体及3'非特异结合等问题。简单的计算公式可以用2+4的公式。若你计算的Tm值达到了快90 ,不包括酶切位点。引物公司给你发的单子是包括酶切位点的。自己可以再估计一下。如你设计了带酶切位点的引物,总长分别为29、33个碱基,去掉酶切位点和保护碱基,分别为17、21个碱基。引物公司给的单子是70多度,实际用的只有50度,用55度扩的结果也差不多。 其它关于Tm值的计算,有用PP5.0进行评价的,需要考虑的参数包括:base number、GC%、Tm、hairpin、dimer、false priming、cross dimer。退一般退火温度为Tm-5度,退火温度的计算可以不把加入的酶切位点及保护碱基考虑进去,如上所言,PCR几个循环后,引物外侧的序列已经参入了扩增片断中,所以你可以在预变性后多加几步,温度比你Tm值低些(这样可能会增加非特异性),Tm值是你包括酶切位点及保护碱基的Primer计算出来的。1.一般在5'端加保护
测序引物设计指南
测序引物设计指南 ?PCR引物设计方法: 1.引物最好在模板cDNA的保守区内设计。 DNA序列的保守区是通过物种间相似序列的比较确定的。在NCBI上搜索不同物种的同一基因,通过序列分析软件(比如DNAman)比对(Alignment),各基因相同的序列就是该基因的保守区。 2.引物长度一般在15~30碱基之间。 引物长度(primerlength)常用的是18-27bp,但不应大于38,因为过长会导致其延伸温度大于74℃,不适于TaqDNA 聚合酶进行反应。 3.引物GC含量在40%~60%之间,Tm值最好接近72℃。 GC含量(composition)过高或过低都不利于引发反应。上下游引物的GC含量不能相差太大。另外,上下游引物的Tm值(meltingtemperature)是寡核苷酸的解链温度,即在一定盐浓度条件下,50%寡核苷酸双链解链的温度。有效启动温度,一般高于Tm值5~10℃。若按公式Tm=4(G+C)+2(A+T)估计引物的Tm值,则有效引物的Tm为55~80℃,其Tm值最好接近72℃以使复性条件最佳。 4.引物3′端要避开密码子的第3位。 如扩增编码区域,引物3′端不要终止于密码子的第3位,因密码子的第3位易发生简并,会影响扩增的特异性与效率。 5.引物3′端不能选择A,最好选择T。 引物3′端错配时,不同碱基引发效率存在着很大的差异,当末位的碱基为A时,即使在错配的情况下,也能有引发链的合成,而当末位链为T时,错配的引发效率大大降低,G和C错配的引发效率介于A、T之间,所以3′端最好选择T。 6.碱基要随机分布。 引物序列在模板内应当没有相似性较高,尤其是3’端相似性较高的序列,否则容易导致错误引发(Falsepriming)。降低引物与模板相似性的一种方法是,引物中四种碱基的分布最好是随机的,不要有聚嘌呤或聚嘧啶的存在。尤其3′端不应超过3个连续的G或C,因这样会使引物在GC富集序列区错误引发。 7.引物自身及引物之间不应存在互补序列。 引物自身不应存在互补序列,否则引物自身会折叠成发夹结构(Hairpin)使引物本身复性。这种二级结构会因空间位阻而影响引物与模板的复性结合。引物自身不能有连续4个碱基的互补。 两引物之间也不应具有互补性,尤其应避免3′端的互补重叠以防止引物二聚体(Dimer与Crossdimer)的形成。引物之间不能有连续4个碱基的互补。 引物二聚体及发夹结构如果不可避免的话,应尽量使其△G值不要过高(应小于4.5kcal/mol)。否则易导致产生引物二聚体带,并且降低引物有效浓度而使PCR反应不能正常进行。
相关文档
- 引物设计心得、Primer5.0 使用
- 酶切位点设计和选择
- 各种酶切位点的保护碱基引物设计必看
- 设计引物如何设计酶切位点
- shRNA设计原则
- 设计引物如何设计酶切位点
- 引物设计原及酶切位点选择和设计
- 设计引物如何设计酶切位点
- 酶切位点的设计
- 引物设计原则及酶切位点选择和设计
- 质粒目的基因插入及引物设计流程
- 酶切位点的设计
- 酶切位点的设计标记
- 引物设计原则及酶切位点选择和设计
- PCR设计引物时酶切位点的保护碱基
- 各种酶切位点的保护碱基(引物设计必看)
- 酶切位点设计和选择
- PCR设计引物时酶切位点的保护
- 实验2-设计性实验-不同限制性内切酶对质粒切割-指导讲解
- 引物设计原则及酶切位点选择与设计